
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Remember me
This paper introduces a novel multimodal optical microscope, integrating Raman and laser-induced photoluminescence (PL) spectroscopy for the analysis of micro-samples relevant in Heritage Science. Micro-samples extracted from artworks, such as paintings, exhibit intricate material compositions characterized by high complexity and spatial heterogeneity, featuring multiple layers of paint that may be also affected by degradation phenomena. Employing a multimodal strategy becomes imperative for a comprehensive understanding of their material composition and condition. The effectiveness of the proposed setup derives from synergistically harnessing the distinct strengths of Raman and laser-induced PL spectroscopy. The capacity to identify various chemical species through the latter technique is enhanced by using multiple excitation wavelengths and two distinct excitation fluence regimes. The combination of the two complementary techniques allows the setup to effectively achieve comprehensive chemical mapping of sample through a raster scanning approach. To attain a competitive overall measurement time, we employ a short integration time for each measurement point. We further propose an analysis protocol rooted in a multivariate approach. Specifically, we employ non-negative matrix factorization as the spectral decomposition method. This enables the identification of spectral endmembers, effectively correlated with specific chemical compounds present in samples. To demonstrate its efficacy in Heritage Science, we present examples involving pigment powder dispersions and stratigraphic micro-samples from paintings. Through these examples, we show how the multimodal approach reinforces material identification and, more importantly, facilitates the extraction of complementary information. This is pivotal as the two optical techniques exhibit sensitivity to different materials. Looking ahead, our method holds potential applications in diverse research fields, including material science and biology.
Multimodal imaging is a rapidly emerging approach that integrates multiple imaging modalities to provide a comprehensive understating of a sample [1]. Specifically, direct correlative multimodal imaging refers to the ability of gathering information on the same specimen using two or more complementary modalities employed sequentially in a hybrid setup [2]. The combination of multiple techniques, each possessing distinct spatial resolutions and sensitivities to various sample properties, both structural and functional, facilitates the acquisition of a composite and complementary view of the sample under investigation. The approach is mainly exploited in microscopy to study biological samples by combining techniques such as Raman spectroscopy, fluorescence lifetime imaging microscopy, optical coherence tomography, second harmonic generation, electron microscopy, and atomic force microscopy [3–6].
Indeed, from a material point of view, artifacts, as painting, sculptures and archaeological objects, are very complex due to the presence of multiple chemical species, including degradation products, heterogeneously distributed from the surface down to inner layers. The analysis of such materials from a scientific perspective is indeed conducted using a wide range of in-situ and laboratory techniques that, given the value of the objects under investigations, are preferably non-destructive and non-invasive. By gathering a set of spectroscopy data and images of the art object, a multimodal protocol is used to map the different artistic materials present in artworks. The approach typically involves the sequential employment of different setups [7, 8], while there are only a few examples where a single setup, mounting different imaging or mapping spectroscopy units, can probe the artwork surface with different spectroscopy techniques [9–11].
In HS this multimodal methodology proves effective also in microscopy to analyze micro-samples taken from works of art. The conventional procedure is to sequentially investigate micro-samples with different techniques, starting from simple optical inspection and then moving to FT-IR, Raman, photoluminescence (PL) and scanning electron microscopies. Nonetheless, the utilization of distinct microscopy setups results in different sample placement, making difficult to accurately align and overlap the multiple sets of data collected. Furthermore, different sample preparation methods, sometimes irreversible, are needed for each technique in the process.
The aim of this work is to present an innovative hybrid microscopy system specifically designed for mapping the surfaces of micro-samples by a raster scanning approach. The instrument combines two optical techniques, Raman and PL spectroscopies, which allows sample investigation in a non-destructive way preserving its integrity. The two methods are highly complementary, and their coupling in a unique hybrid set-up as a point-analysis technique has been already proved effective [12–14]. In HS, Raman micro-spectroscopy is widely used for material identification, with specific reference to artist pigments, thanks to its high chemical sensitivity [15–17]. On its turn, the effectiveness of micro-PL spectroscopy in HS is limited by the impossibility of using fluorescent markers to target materials, as it happens in the molecular biology. Despite this, the technique has proved valuable to identify lake pigments, organic colorants, and luminescent semiconductor pigments [18, 19], especially when combined with high spatial and spectral resolution [20] and/or time-resolved capabilities [21, 22]. The effectiveness of PL spectroscopy for material identification in HS is further augmented by its combination with the reconstruction of historical materials and chemometrics [21–24]. The developed system aims to be an innovative setup specifically tailored for surface chemical mapping of samples. When dealing, for instance, with stratigraphic micro-samples taken from paintings it is crucial to detect the distinct spatial arrangements of paint layers and of neo-formation degradation products that can be formed at the sample surface or at the interface between layers. By providing a detailed spatial information through mapping, the developed system enhances our ability to understand the chemical composition and morphology of samples, facilitating a more comprehensive and insightful analysis.
In this study, after a detailed description of the multimodal mapping microscope, we introduce a specialized analysis protocol crafted to extract insightful information from the collected hyperspectral mapping datasets. To discern pertinent details about the chemical species and their spatial distribution throughout the sample, we employed a multivariate analysis protocol grounded in spectral decomposition. This approach identifies spectral endmembers and breaks down each spectrum into a linear combination of these components, weighted according to their specific abundance. Principal component analysis (PCA) is the most widely used technique for spectral decomposition, but it has notable limitations. It focuses on finding linear combinations of spectral features that capture the maximum variance in the dataset, lacking direct physical interpretation of the principal components as they are abstract directions in the original feature space determined by eigenvectors. Further, it generates principal components with both positive and negative values, complicating the assignment of a clear and intuitive physical meaning to each component.
Instead, we turn to non-negative matrix factorization (NMF) [25], a decomposition method that ensures non-negativity in both endmembers and abundances, facilitating a direct and physically interpretable understanding of the spectral components. Specifically, in the current investigation we analyze the gathered hyperspectral mapping datasets using NMF, enhancing its performance by incorporating non-negative double single value decomposition (NNDSVD) as the initialization method [26], as will be detailed in the methodological section.
2.1. Design of the instrumentThe developed setup employs different excitation paths that focus the beam on a micrometric spot (as detailed in table 1) on the sample surface, and a single collection path to retrieve the emitted or scattered signal from the materials. The setup works on the same principle of a simple scanning confocal microscope. As such it could theoretically achieve sub-micrometer resolution in the lateral direction, and micrometer-scale resolution in the axial direction. However, due to the long integration time required by Raman spectroscopy, diffraction limited imaging would only be possible by spatially scanning an extremely limited field of view in the experimental time constrains. The resolution of the system is therefore intentionally limited by underfilling the objective's aperture with the illumination sources and by employing a multi-mode fiber in the collection path as a non-diffraction-limited pinhole. This both increases the sample area covered by one image pixel, and greatly increases the photon flux at the detector for a given illumination power at the sample, therefore enabling mapping of a field of view of hundreds of micrometers in lateral size in a reasonable time. Moreover, the wide fiber used as a pinhole increases the axial extent of the point spread function, compensating for the non-planar nature of the samples considered.
Table 1. Specifications. Table summarizing the spatial resolution of the setup—achievable with the two different objectives (10X, 20X) and different excitation wavelengths and the typical power density on sample.
ObjectiveExcitation wavelength (nm)Excitation spot diameter size (μm)Collection spot diameter size (μm)Average power on sample (mW)Average power density on sample (kW cm−2)10X 35520500.350.1137510501.11.1678522504.21.1020X 35510250.150.193755250.562.8578511254.14.31In addition to the scanning system, a wide-field, white-light imaging path has been implemented for quick selection of the subarea of interest. The general scheme of the setup is presented in figure 1.
Figure 1. Setup. (A) Layout of the microscopy setup with the three laser excitation paths, the optical path for sample illumination and visualization and the Raman and PL collection path. CL = Clean Laser, BS = Beam Splitter, HP = High Pass. (B) Example of a painting with its sampling point and (C) the corresponding stratigraphic micro-sample to be analyzed. (D) Photography of the developed setup.
Download figure:
Standard image High-resolution imageThree laser sources have been employed to stimulate Raman and PL emissions of the materials:
A continuous-wave (CW) compact diode laser (TEC-510-0785-200 Lion series, Sacher Lasrtechnik), with an emission wavelength of 785 nm and a narrow spectral width of 300 kHz (10−5 cm−1) for Raman spectroscopy. The beam is coupled with the setup through a mono-mode fiber with a core diameter of 5 μm, whose output beam is then collimated by a collimator with 4.51 mm focal length (F230FC-780).A diode pumped Q-switched solid-state Nd:YAG laser (FTSS 355–50-STA, CryLaS) with third harmonic emission at 355 nm. The laser radiation is characterized by a repetition rate of 100 Hz and a pulse duration of 1 ns. Each pulse carries an energy of 70 μJ and the average output power is 7 mW.A CW diode laser (CL 2000, CrystaLaser) emitting at 375 nm, with average output power of 5 mW. It is positioned along the same optical path of the 355 nm excitation.The two latter sources, used for exciting the PL emission, are coupled to the microscope through free-beam configuration, and a moveable mirror allows one to switch between them. It must be noted that these two laser sources, although having close excitation wavelength and similar average power, have very different peak powers, with the pulsed laser source providing a peak power that is 7 orders of magnitude larger than the power of the CW laser. As already demonstrated in past studies, the different working regimes of the two laser sources allow one to probe different recombination paths in crystalline materials, even when collecting the emission with a steady-state detector [27]. In particular, the Q-switched laser provides a high number of photons in the nanosecond pulse time. Consequently, upon huge excitation of conduction band states, the saturation of the strongly coupled trap state (TS) occurs, thus promoting the emission through other recombination paths, typically near-band edge (NBE) radiative transitions. Conversely, the CW laser provides the same average number of photons to the sample, but distributed over time. Hence, generally it does not give rise to saturation of TS states and allows the detection of the emission occurring from these energetic levels.
The three excitation lights, aligned along the same optical path within the microscope, pass through a filter wheel and are focused on sample through a proper objective. The filter wheel houses dichroic mirrors at 785 nm (Semrock, LPD02-785RU) and 389 nm (Semrock, FF389-Di01) and a 50:50 beam splitter. The latter is used to illuminate the field of view with a halogen lamp, as described later, to image the sample and identify the area to be analyzed. The detection path includes, apart for the already quoted objective and dichroic mirror, an interchangeable high pass filter (LP02-785RE-25 for Raman and different Thorlabs FELHs for fluorescence) to remove the residual laser light and an achromatic lens with 20 mm focal length and a coating for the range 400–1100 nm, to project the image of the analyzed spot onto the tip of a collection fiber, with a core diameter of 50 μm. This enables the effective collection of both PL and near-IR Raman signal, considering their very different spectral range. The core of the collection fiber affects the spectral resolution, as it acts as the slit of the spectrometer. Therefore, the selected core size represents a compromise between achieving efficient signal collection and maintaining a good spectral resolution. The distal tip of the collection fiber is coupled to the spectroscopy detection unit, made of a spectrometer (Acton, SpectraPro-300i, focal length = 300 mm) and a cooled CCD camera (Andor, iDus DV 401). The spectrometer mounts two different gratings (600 lpmm and 300 lpmm) that can be interchanged to achieve the required spectral resolution for resolving, respectively, Raman peaks and PL bands. Specifically, the spectral resolution is equal to 9.8 cm−1 for Raman measurements, and 1.9 nm for PL ones.
In terms of spectral range covered, the ultimate lower limit of detectable Raman peaks is set by the spectral transmission of the dichroic and long pass filters, with the dichroic filter having the most limiting spectral specifications: it ensures high transmission (>80%) from 793 nm, equivalent to a minimum Raman shift of ∼130 cm−1. However, in the current research the grating has been centered at 910 nm and, considering the calibration of the spectrometer, it allows the investigation of the spectral range from 208 cm−1–2752 cm−1. On turn, the spectral range covered for PL analysis spans from 366 nm to 876 nm.
Two further optical paths are implemented to allow the illumination and visualization of the sample. A Tungsten Halogen lamp (Ocean Insight, HL2000) is coupled to a large core (400 μm) optical fiber whose distal end is imaged to the object plane through a 4-lens optical system, which includes the microscope objective. This provides a uniform illumination to the sample, without the need of a condenser. Details of the design of this optical system simulated via the Ansys Zemax ray-tracing software is provided in supporting information. The sample is then imaged by a color camera through a simple tube lens.
To achieve the desired spatial resolutions depending on the analyzed sample, different objectives can be employed, typically a 10X (NA = 0.25) or a 20X (NA = 0.4). The setup specification achieved with these two objectives, as power density on sample and excitation and analysis lateral spot size, are listed in table 1. The axial resolution is limited by the collection fiber size at approximately 180 μm for the 20X objective and 280 μm for the 10X objective.
2.2. Data post-processing and analysisThe acquisition protocol consists of obtaining at least two hyperspectral datacubes of the same area on the sample by sequentially employing the different excitation sources. The single-point integration time for each measurement is chosen to achieve a competitive total acquisition time, ensuring that a hypercube acquisition would not be longer than 2 h.
Once datasets are collected, the analysis protocol for each dataset applies a standard spectral pre-processing that includes spike cleaning, smoothing, median filtering and, in case of Raman data, baseline removal using the SNIP clipping algorithm [28].
After pre-processing, data analysis is aimed at spectral decomposition. Each pixel of a spectral datacube contains a Raman or PL spectrum that are a combination of multiple spectra originating from different compounds present in the analysis point. Consequently, spectral decomposition is the method of choice for extracting the spectral components present in the acquired dataset and their abundances in the two-dimensional space [29].
For the purpose, the hyperspectral datacube is rearranged in a data matrix  made of
made of  pixels, while
pixels, while  is the spectral dimension. The data matrix is then factorized as the product of
is the spectral dimension. The data matrix is then factorized as the product of  spectral endmembers represented by the matrix
spectral endmembers represented by the matrix  and the abundance matrix
and the abundance matrix  , which represents the contribution of each endmember to a given pixel [30]. The process is summarized as follows:
, which represents the contribution of each endmember to a given pixel [30]. The process is summarized as follows:
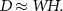
NMF [25] is here applied to achieve spectral decomposition. To reach a stable or optimal solution, convergence is crucial as it indicates that further iterations will not significantly improve the factorization outcome. The most used convergence method is through Euclidean norm. However, due to the non-convex nature of the NMF problem, finding the global optimal solution is generally not guaranteed and multiple local minima are present. The convergence can be significantly improved through the implementation of an appropriate initialization mechanism, to achieve efficient factorization results [29, 31–33]. For the purpose, in this paper we employ the NNDSVD initialization method [26], which is based on a two SVD processes, to obtain the initial values for the endmember matrix W and the abundance matrix H. Initially, a truncated SVD is used to estimate the data matrix D. Next, unit rank matrices are constructed from the pairs of singular vectors previously obtained. Subsequently, the positive components of these unit rank matrices are isolated, and a secondary SVD is performed on them to extract singular triplets. These extracted triplets are employed to initialize matrices W and H in NMF. By avoiding random initialization for matrices H and W, the approach prevents the convergence to local minima, ensuring consistent convergence to the same solution across multiple runs and rapidly reducing the approximation error. Moreover, through the utilization of the NNDSVD initialization method, the NMF algorithm starts with initial values that strictly adhere to the nonnegativity constraint.
The analysis protocol, comprising preprocessing and spectral decomposition is provided in figure 2.
Figure 2. Analysis protocol. (A) Block diagram of preprocessing and analysis protocol, starting from the two raw hypercubes from Raman and PL acquisition and giving as output the Raman and PL endmembers and abundance maps. (B)–(E) An example of the protocol: (B) raw Raman spectrum of a single pixel, (C) pre-processed spectrum of the same pixel, (D) extracted spectral endmembers and (E) abundance maps provided as a false RGB representation.
Download figure:
Standard image High-resolution imageThe analyzed samples include dispersions of mixture of pigments and two stratigraphic micro-samples taken from paintings. Results of the analysis of a third paint stratigraphy is provided in supporting information.
The first two samples are mixtures of pigment powders. The first one is a dispersion of two polymorphs of Titanium White, namely rutile (Kronos® 2900) and anatase (Kronos® 1002) as 1:1 weight proportion [34, 35]. The second one is a dispersion containing a combination of white pigments, namely zinc white (ZnO, Kremer®), gypsum (CaSO4 + 2H2O) and rutile (polymorph of TiO2, Kronos® 2900) in proportions of equal weight.
The last samples are two micro-samples taken from paintings and prepared as polished cross-section to reveal the painting stratigraphy. The first one originates from Michail Larionov's painting 'Raggism', a vibrant painting of the early 20th century. Supporting information provides an overview of the artwork, along with an image of the sample, labeled as L5, prepared as a stratigraphic cross-section with multiple paint layers. The stratigraphy (figure 3(a)) comprises, from top to bottom, a yellow layer featuring green stripes, a white layer containing an orange insert and a light blue layer, mixed with orange [22, 36]. The second paint stratigraphy comes from Natal'ja Gončarova's painting titled 'Raggism Forest', which dates back to the second decade of 1900. The supporting information provides an overview of the artwork, accompanied by an image of the sample, labeled as G1. This sample reveals multiple layers, comprising three distinct water green layers on the top, a big white central layer, and a lower blue layer.
Figure 3. Analysis of TiO2 powder. (A) Visible picture of TiO2 dispersion with the closed red line indicating the rectangular area mapped with the multimodal setup. (B) and (D) Composite images created by merging the abundance map of rutile (red) and anatase (green), identified through (B) Raman and (D) PL. The corresponding spectral endmembers are provided in (C) and (E), with indication of Raman shifts and shadowed areas the PL bands that allows for the identification of two TiO2 polymorphs.
Download figure:
Standard image High-resolution image 4.1. Mixture of two polymorphs of the same pigmentThe powder dispersion of a mixture of two TiO2 polymorphs were analyzed with the 10X objective, scanning an area of 500 μm × 600 μm in lateral size with a step size of 10 μm. Each data point was acquired with an integration time of 1 s, resulting in a total acquisition time of approximately 50 min for each spectral dataset. Raman acquisition utilized a 785 nm excitation wavelength, whereas for PL only the CW laser at 375 nm was employed.
The two polymorphs could be detected with both Raman and PL spectroscopy mapping, as shown in figure 3. Specifically, two Raman spectral endmembers are extracted, related to rutile (Raman shifts at 445 and 620 cm−1) and anatase (Raman shifts at 404, 525, 650 cm−1). Similarly, the two extracted PL spectral endmembers show the emission spectral features of rutile (TS emission band centered at 810 nm) and anatase (emission band centered around 480 nm) [27, 37].
4.2. Mixture of different white pigmentsFor the analysis of the mixture of different white pigments, a 10X objective was employed, and an area of 400 μm × 600 μm was mapped with a step size of 10 μm. The acquisition time for each data point was set at 1 s, leading to a total measurement time of about 40 min for each dataset. Raman spectroscopy was performed with 785 nm excitation, while for PL, both pulsed and CW excitations were employed.
In figure 4, Raman and PL spectral endmembers are displayed along with their corresponding abundance maps. Specifically, Raman spectral endmembers make it possible to identify rutile (Raman shifts at 445 and 620 cm−1) and gypsum (Raman shifts at 1023 and 1150 cm−1). PL spectral end-members vary depending on the employed excitation peak power: the high-peak power excitation allows the identification of zinc white (ZnO) through the tail of its NBE PL emission centered at 380 nm; the low-peak power excitation still allows the identification of zinc white, but in this case through its TS PL emission band centered at around 530 nm, and of rutile through its TS emission centered at around 810 nm [27, 37].
Figure 4. Analysis of mixture of white pigments. (A) Visible picture of the mixtures of white pigments with the closed red line indicating the rectangular area mapped with the multimodal setup. (B) Composite image created by merging the abundance map of the two Raman spectral endmembers provided in (C) with indication of the Raman shifts that allows for the identification of rutile (red) and gypsum (green). (D) Abundance map of the PL spectral endmember (following excitation by the pulsed laser) provided in (E) and resembling the tail of Zinc White bandgap emission. (F) Composite image created by merging the abundance map of the two PL spectral end-members (following excitation with the CW laser) provided in (G) where shadowed areas highlight the PL bands shifts that allows for the identification of rutile (red) and Zinc White (blue, trap state emission).
Download figure:
Standard image High-resolution image 4.3. Stratigraphic micro-sample: Larionov L5Raman and PL measurements, excited at 785 nm and 355 nm respectively were carried out to identify artist pigments within the layers of the paint stratigraphy. The acquired maps of the sample were obtained using a 20X objective, covering dimensions of 300 μm × 350 μm, with a step size of 5 μm. Each measurement point required 1 s acquisition for a total measurement time of about 1 h and 10 min for each hypercube.
Raman spectroscopy and following spectral data decomposition allowed the extraction of four spectral endmembers ascribable to the presence of four different artist pigments, in some sample areas present as pure pigments, in other as pigment mixtures. Specifically, the four spectral endmembers resemble the Raman spectral features of (i) chrome yellow (PbCrO4, detected Raman shifts at 351 and 844 cm−1), identified in the yellow layer, (ii) phthalocyanine blue (C32H16CuN8, detected Raman shifts at 687, 821, 856, 1219, 1294, 1346 and 1535 cm−1), identified in some subareas of the yellow layer, (iii) ultramarine blue (Na8-10Al6Si6O24S2-4, detected Raman shifts at 546 cm−1), identified in the blue layer, and (iv) cinnabar (HgS, detected Raman shifts at 245 and 340 cm−1), identified in specific reddish points within the white layer. Raman findings are summarized in figures 5(b) and (c) and in table 2.
Figure 5. Analysis of L5 paint stratigraphy. (A) Visible picture of the paint stratigraphy with the dashed closed white line indicating the rectangular area mapped with the multimodal setup. (B) Composite image created by merging the abundance map of the four Raman spectral endmembers provided in (C) with indications of the Raman shifts that allows for the identification of Cinnabar, phthalocyanine blue, ultramarine blue and chrome yellow. (D) Composite image created by merging the abundance map of the three PL spectral endmembers provided in (E) where the shadowed area highlights the PL bands that allows for the identification of zinc white, cadmium yellow, and cadmium orange (F) summary of the findings achieved with the multimodal microscopy set-up in terms of abundance maps of all the identified pigments superimposed to the color picture of the paint stratigraphy.
Download figure:
Standard image High-resolution imageTable 2. L5 Peaks. Identified peaks of both Raman and PL maps in sample L5 along with their pigment attribution.
Identified pigment (chemical formula)Identification throughRaman shifts (cm−1)Wavelength of PL emission peak (nm)Cinnabar (HgS)Raman245, 340—Chrome yellow (PbCrO4)Raman351, 844—Ultramarine blue (Na8-10Al6Si6O24S2-4)Raman546—Phthalocyanine blue (C32H15CuN8)Raman687, 821, 856, 1219, 1294, 1346, 1535—Zinc white (ZnO)PL—400 (tail)Cadmium yellow light ((Cd, Zn)S)PL—500Cadmium orange (Cd(S,Se))PL—550Mapping with PL spectroscopy allowed us to extract complementary information that enabled us to identify other pigments in the paint layers. Specifically, the spectral unmixing procedure allowed the identification of three spectral endmembers with spectral features resembling the ones of (i) zinc white (ZnO), identified in the white layer and distinguished by the tail of its (NBE PL emission at 380 nm [38]), (ii) cadmium orange (Cd(S,Se)), identified in the reddish stripe within the blue layer and distinguished through its NBE emission at 550 nm [39], and (iii) cadmium yellow light ((Cd,Zn)S), identified in the yellow layer and distinguished through its characteristic NBE emission at 500 nm [39]. PL findings are summarized in figures 5(d) and (e) and in table 2.
4.4. Stratigraphic micro-sample: Goncharova G1Raman and PL measurements were conducted to identify the pigments, with excitation laser sources at 785 nm and 355 nm respectively. The acquired maps of the sample were obtained using a 20X objective, covering an area of 150 μm × 500 μm, with a step size of 5 μm. Each measurement point was acquired with 1 s exposure time, resulting in a total measurement time of approximately 1 h for each hypercube.
Through Raman spectroscopy and subsequent NMF analysis, five distinct spectral endmembers corresponding to different pigments were identified. These included: (i) Cinnabar (HgS), with detected Raman shifts at 245 and 340 cm−1, identified in a small stripe in the top part of the sample. (ii) Phthalocyanine green (C32H16CuN8), with detected Raman shifts at 683, 739, 775, 1218, 1291, 1345, and 1542 cm−1, identified in two stripes in the top part. (iii) Ultramarine blue (Na8-10Al6Si6O24S2-4), with detected Raman shifts at 545 cm−1, identified in the lower blue part. (iv) Prussian blue (C18Fe7N18), detected by Raman shifts at 528 cm−1 and 2125 cm−1 and enclosed in the two phthalocyanine stripes in the upper part. (v) Zinc white (ZnO), with detected Raman shifts at 425 cm−1, identified in the entire white part of the sample. These Raman identifications are summarized in figures 6(b) and (c).
Figure 6. Analysis of G1 paint stratigraphy. (A) Visible picture of the stratigraphic micro-sample with the dashed rectangular white line indicating the area mapped with the multimodal setup. (B) Composite image created by merging the abundance map of the four Raman spectral endmembers provided in (C) with indications of the Raman shifts that allows for the identification of cinnabar, phthalocyanine, ultramarine blue, prussian blue and zinc white. (D) Composite image created by merging the abundance map of the three PL spectral endmembers provided in (E) where the shadowed areas highlight the PL bands that allows for the identification of zinc white and cadmium yellow.
Download figure:
Standard image High-resolution imageAdditionally, PL spectroscopy mapping provided complementary insights: the data decomposition unveiled two spectral endmembers corresponding to: (i) Zinc white (ZnO), previously identified with Raman spectroscopy in the white layer, distinguished by the tail of its NBE PL emission at 380 nm. (ii) Cadmium yellow light ((Cd,Zn)S), identified in the water green layer, mixed with ultramarine blue, and distinguished through its characteristic NBE emission at around 500 nm. The PL spectra are shown in figures 6(d) and (e).
The results obtained from the analysis of these samples underscore the remarkable effectiveness of the multimodal approach for material identification. This approach not only provides complementary information through PL and Raman spectroscopy, but also enhances the robustness of material identification, as is discussed below.
In the examination of titanium dioxide powder dispersion, both Raman and PL acted successfully to discriminate between the two polymorphs and to reconstruct their spatial distribution, with some sample areas displaying the presence of one polymorph only, and other areas displaying their co-presence.
Moving to a more complex sample, that is the mixture of different white pigments, the combination of Raman and PL techniques on one hand strengthens the identification of a specific material, rutile, identified by both techniques. On the other hand, it also yields complementary information, with gypsum and zinc white identified by Raman and PL spectroscopy, respectively. This example also highlights the efficacy of employing different excitation peak powers for PL measurements, leading to the robust identification of zinc white through the detection of both its NBE and TS radiative recombination, probed respectively through high-peak power and low power excitation.
Finally, the analysis of paint stratigraphies demonstrates how Raman and PL spectroscopies offer remarkably distinct and complementary insights. For what concerns sample L5, Raman spectroscopy proves valuable in deducing the chemical composition of the yellow layer, its greenish stripes, the blue layer at the stratigraphy's base, and reddish points within the white layer. However, it falls short in providing information about the white pigment in the white layer. On the other hand, PL spectroscopy effectively discerns the chemical composition of the white layer, identifies materials in the thin orange layer at the base, and indicates the presence of an additional yellow pigment in the yellow layer. Likewise, sample G3 was entirely identified through the complementary use of both techniques. Specifically, the integration of Raman and PL spectroscopy enabled us to discern subtle differences in color among the first three water green layers. The first layer solely contains phthalocyanine, while the second layer comprises a blend of Prussian blue and zinc white. The third layer consists of a mixture of light chrome yellow and phthalocyanine green.
Multimodality proves highly advantageous when dealing with paint materials containing a strongly fluorescent binder. This challenge is exemplified in the analysis of the two stratigraphic micro-samples, where the presence of zinc white (in sample L5) and cadmium yellow (in both samples) could not be achieved via Raman spectroscopy due to their Raman signals being obscured by the intense background fluorescence of the binder matrix embedding the pigments. Despite partially mitigating fluorescence emission from organic compounds by employing a near-infrared source to excite Raman signals, the binder's fluorescence often remained dominant, overshadowing faint Raman signals. In such instances, PL spectroscopy became essential for identifying these pigments through the detection of their distinct NBE emissions. It is noteworthy that both cadmium yellow and zinc white exhibit minimal Raman signals under 785 nm excitation, with their Raman signals being better stimulated using green excitation [38–41]. Note that the significant impact of the binder's optical emission on the Raman signals is clearly illustrated by the divergent outcomes in the identification of zinc white in the two paint stratigraphies: while the Raman peaks of this pigment remained elusive in sample L5, they were discernible in sample G1 owing to the comparatively reduced fluorescence intensity of the binder.
A further important feature of the developed set-up is its capability to collect a spectroscopy dataset with high degree of redundancy, because of the recurrence of the same spectral components in many points of the sample
Comments (0)