
Remember me

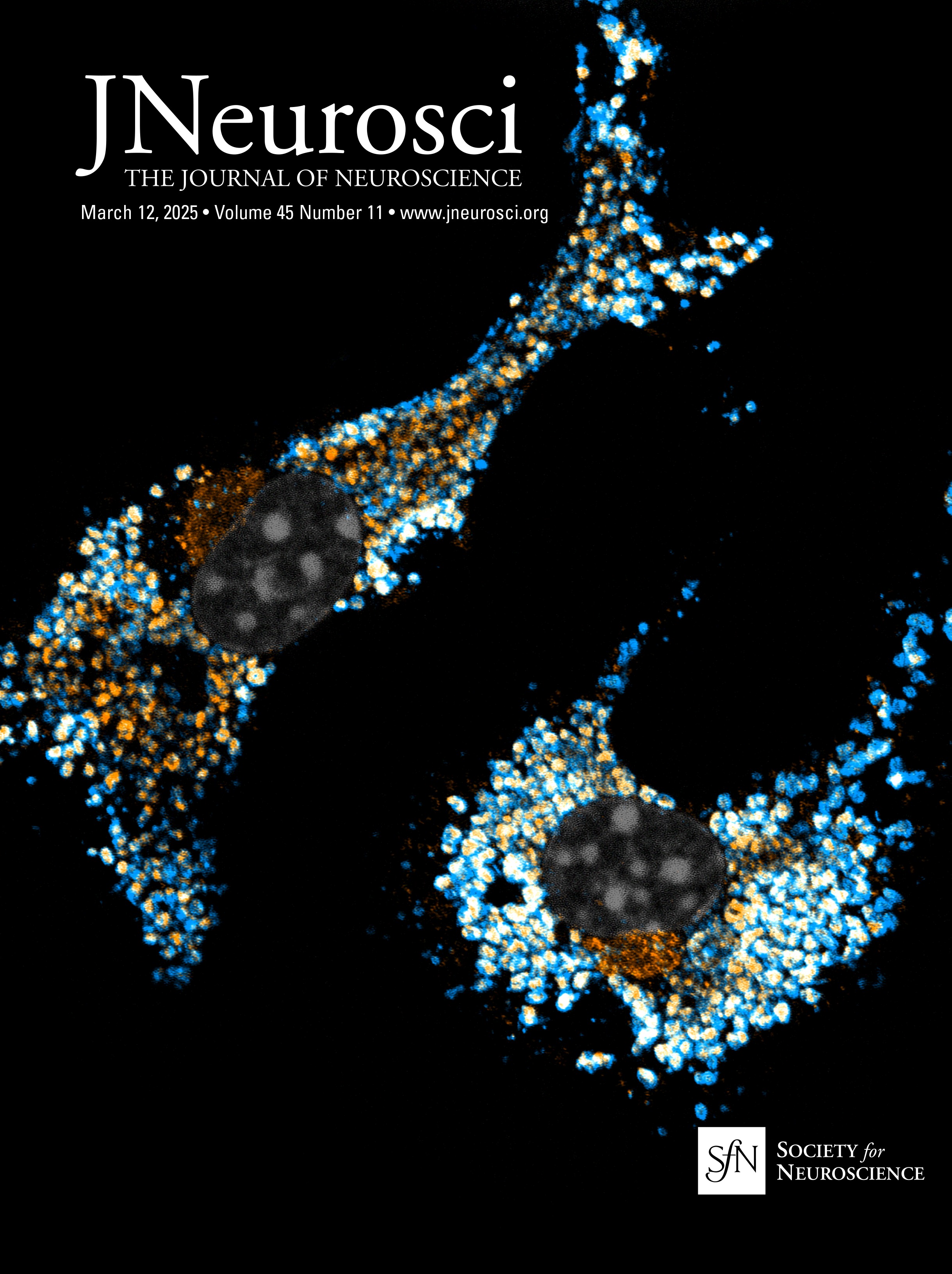
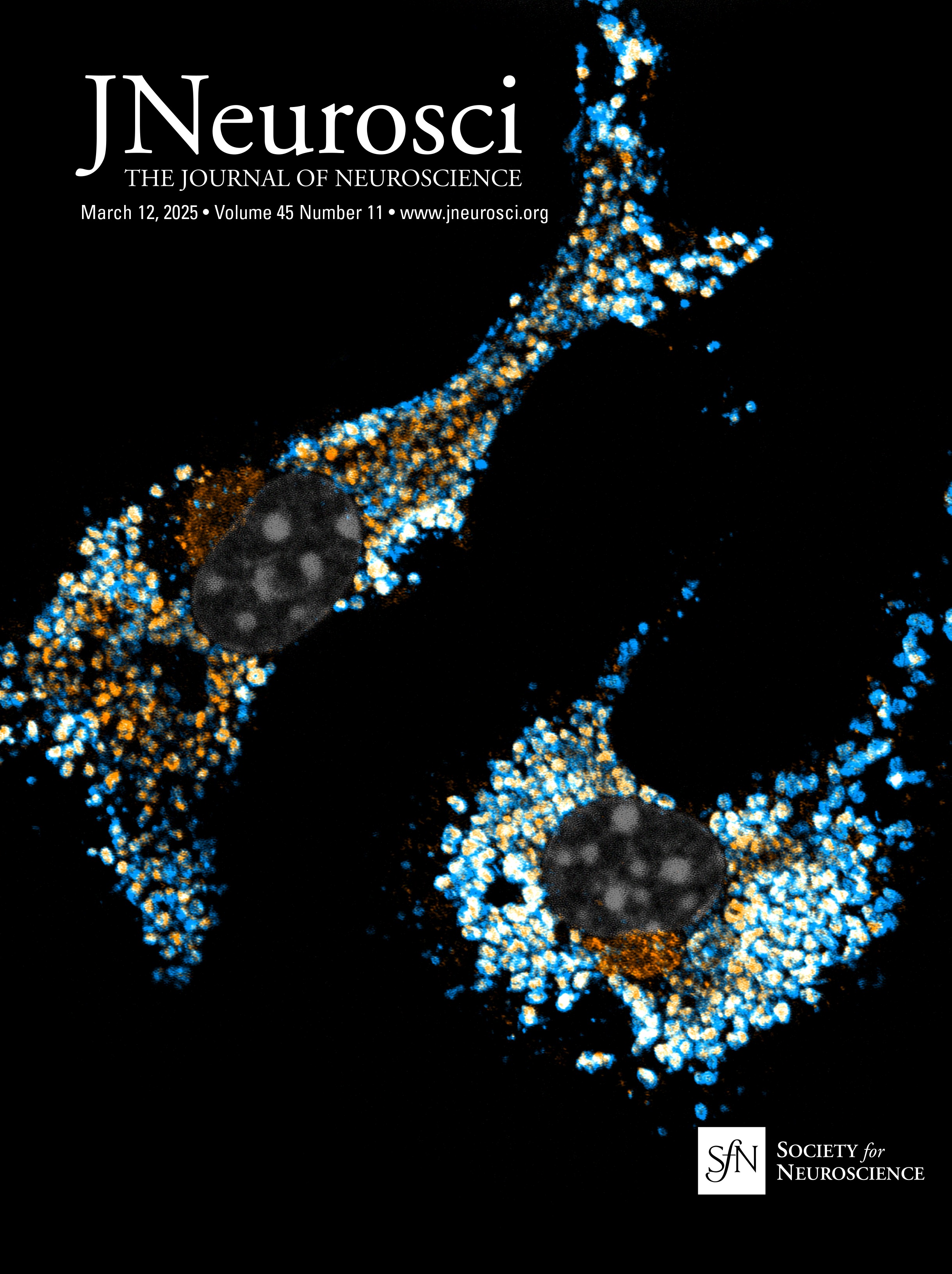



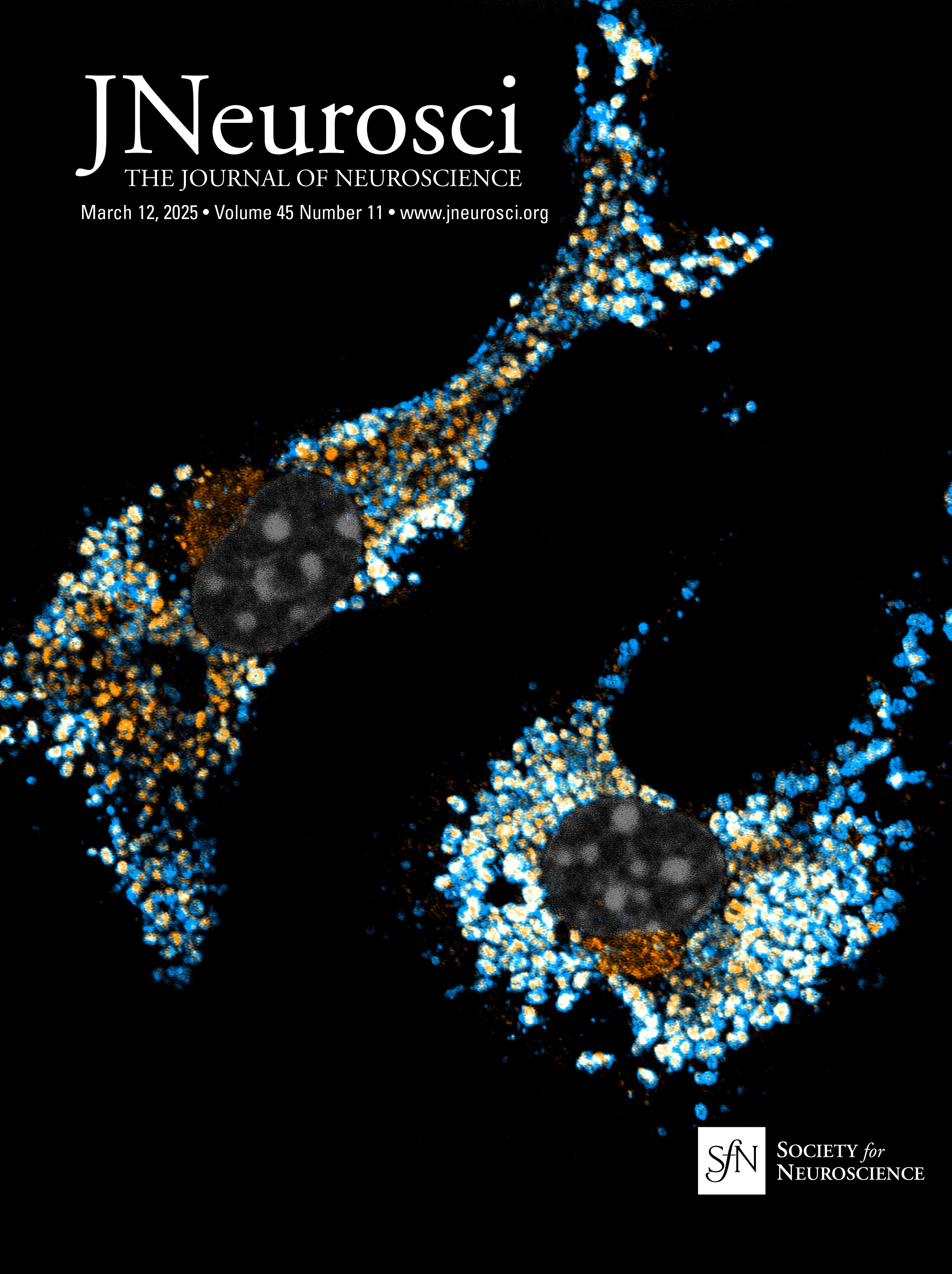
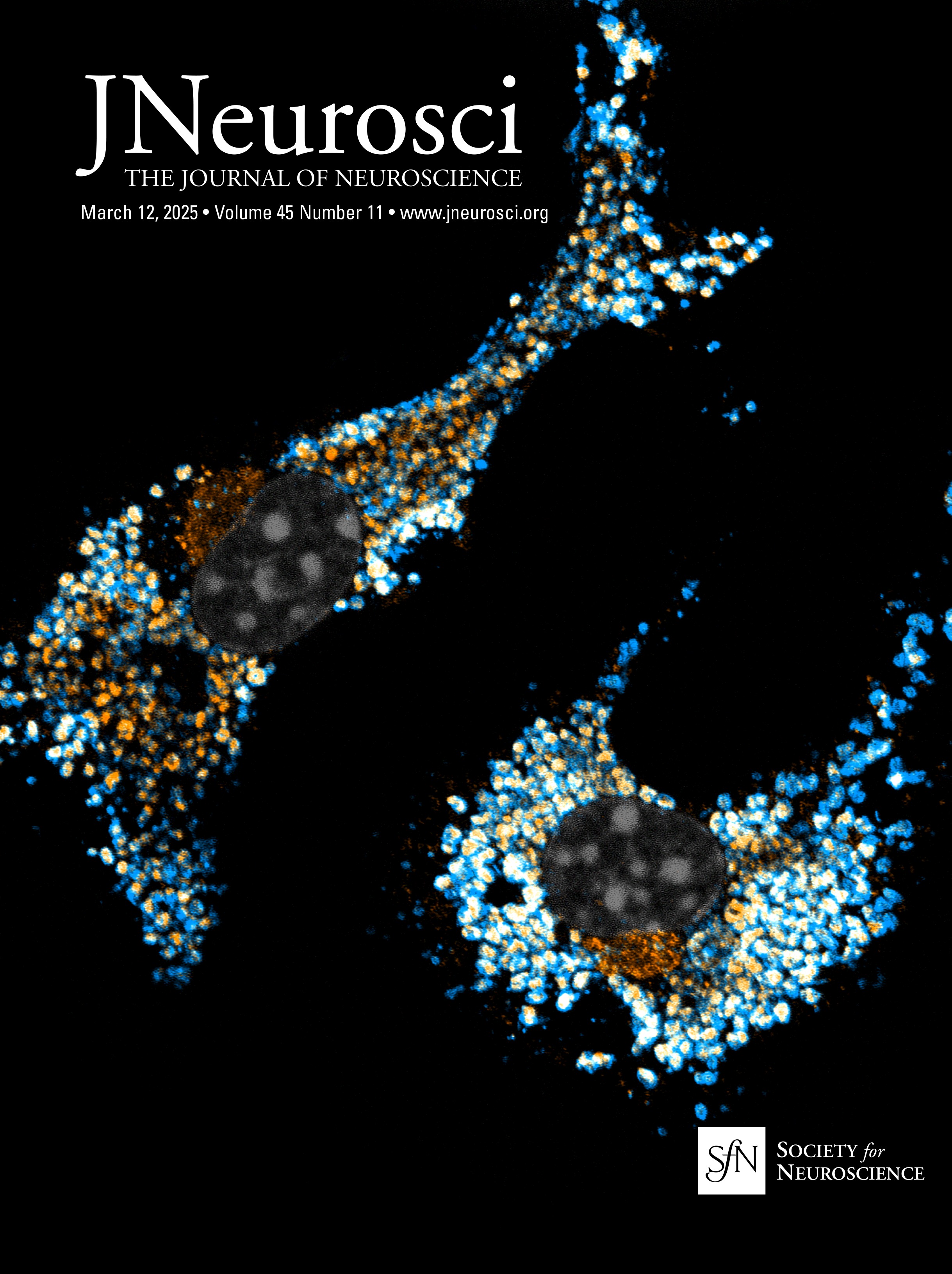

Nrg1 proteins can back signal through at least two different mechanisms: (1) local activation of PI3K-Akt signaling and (2) nuclear translocation of a free C-terminal fragment generated by γ-secretase cleavage. The V321L mutation disrupts the preferred γ-secretase cleavage site (Fig. 1A). To examine the effect of the V321L substitution on Nrg1 signaling, we generated a knock-in germline mutation in mice. The V321L mutation (gtg→ttg) was introduced into C57Bl6 embryonic stem cells using a bacterial artificial chromosome (BAC) construct (Fig. 1B–D). Heterozygotes were interbred, and offspring were born at the expected Mendelian ratios (Fig. 1D). Homozygous V321L mutants were viable and fertile, with no outward morphological or growth abnormalities. Homozygous animals were able to interbreed and yielded normal-sized litters with offspring that appeared healthy. The V321L mutation was also viable on a Type III Nrg1 knock-down background, as a Nrg1L/L × Type III Nrg1+/− cross yielded both Nrg1V/L and Nrg1L/− offspring.
 Figure 1.
Figure 1. Generation of Nrg1 V321L knock-in mice. A, A schematic of the different modes of signaling engaged by Type III Nrg1. Cleavage by Bace1 is an essential step to generate a substrate for γ-secretase. The extracellular EGF-like domain of Nrg1 can interact with ErbB4 on neighboring cells to engage the canonical forward signaling by activating ErbB4. ErbB4 interaction can also result in activation of back signaling by Nrg1. Local back signaling engages PI3K signaling at the membrane, whereas cleavage by γ-secretase results in liberation of the C-terminal ICD, which can translocate to the nucleus–nuclear back signaling. The V321L mutation blocks cleavage by γ-secretase and is predicted to disrupt nuclear back signaling. B, A schematic diagram of the Nrg1 genomic structure. The Nrg1 gene encodes six families of isoforms as a result of alternative promoter usage. Types 1, 2, 4, and 5 contain Ig-like domains (encoded by Exons 3 and 4). Exon 7 is the unique 5′ coding exon for Type III Nrg1, encoding an N-terminal, cysteine-rich transmembrane domain. Exon 8, in combination with Exon 9 or 10, and various combinations of Exons 11, 12, and 13 encode an EGF-like domain common to all Nrg1 isoforms. Exon 13 encodes a common C-terminal transmembrane domain. The C-terminal ICD is encoded by Exons 14–17. The missense SNP that results in a valine-to-leucine substitution in the C-terminal transmembrane domain is shown above the gene. b′, Schematic diagram of the BAC clone used for generating the target construct corresponding to a region of the Nrg1 gene that comprises the entire Type III coding region. Below the BAC clone is a diagram of the targeting construct, including a 5.8 kbp left homology arm, a neoR cassette in the antisense orientation flanked by Frt sites, and a 2.5 kbp right homology arm. b″, Diagram of WT and mutant alleles (the mutant allele following flippase removal of the neo cassette). The C-terminal transmembrane domain sequence is underlined. Black arrows labeled “NDEL1” and “NDEL2” indicate approximate genomic locations of genotyping primers. C, An example genotyping gel of offspring from a het × het cross illustrating a WT, heterozygote and homozygote. D, The mutant mouse line was maintained by breeding heterozygotes. The genotype of pups from >100 litters was analyzed for deviation from the expected 1:2:1 ratio. No deviation was found. Mice with the genotype V/V are referred to as WT in the manuscript and L/L are referred to as V321L as denoted in parenthesis.
Neurons from Nrg1 V321L mutant mice show diminished nuclear back signaling and lack of dendritic growth in response to stimulation with soluble ErbB4The V321L mutation in Nrg1 impairs γ-secretase–mediated cleavage in vitro (Fleck et al., 2016). We performed subcellular fractionation to isolate nuclei and membrane fractions from cortical and hippocampal homogenates of V321L homozygous mutant and their WT litter- and cage mates and compared the levels of Nrg1 ICD by immunoblot analysis (Fig. 2A,B). Nuclei isolated from V321L animals showed lower levels of Nrg1 ICD compared with nuclei from WT counterparts (Fig. 2A). Concurrently, we found higher levels of FL and the membrane-bound ICD (TM-ICD) in the membrane fraction of V321L mice compared with that of WT mice (Fig. 2B). These findings indicate diminished Nrg1 nuclear back signaling in the mutant cortex and hippocampus. We next assessed stimulus-induced nuclear back signaling by the addition of soluble ectodomain of the Nrg1 receptor, Erbb4 (sB4), to cultured hippocampal neurons.
 Figure 2.
Figure 2. The V321L substitution decreases nuclear back signaling. A, Immunoblot of triplicate nuclear fractions isolated from pooled cortical and hippocampal lysates. Nrg1 ICD was detected using Santa Cruz Biotechnology antibody sc-348. Histone H3 served as a nuclear loading control and Na+/K+ ATPase as a marker for the membrane fraction (N = 3 mice/genotype). B, Immunoblot of two replicates of membrane fractions isolated from pooled cortical and hippocampal lysates. NRG1 ICD was detected using Santa Cruz Biotechnology antibody sc-348. Na+/K+ ATPase served as a marker for the membrane fraction; note the lack of the nuclear marker Histone H3 or cytoplasmic marker CyclophilinA indicating clean membrane preps. FL Nrg1 is indicated with a yellow arrowhead as “Nrg1-FL,” and the membrane-bound C-terminal fragment not cleaved by γ-secretase is indicated as “Nrg1 TM-CTF.” A positive control consisting of total lysate from N2A cells transfected with a Type III Nrg1 plasmid is shown in the lane labeled “C.” (N = 2 mice/genotype.) C, Left, Hippocampal neurons from WT (dark blue) and V321L (light blue) neonatal pups (P4) were cultured for 17 d in vitro and were stimulated with either vehicle (Veh), 20 nM sERBB4 (sB4), or 20 nM sErbB4 after a 24 h pretreatment with 20 µM of the γ-secretase inhibitor DAPT (DAPT). Neurons were fixed and stained using an antibody directed against the Nrg1 ICD and counterstained with DAPI. Scale bar, 10 µm. Right, Quantification of nuclear clusters of Nrg1-ICD. Neurons from WT mice show increased nuclear ICD clusters in response to sB4 stimulation, which is counteracted by pretreatment with DAPT (DAPT). Neurons from V321L mice do not respond to sB4 stimulation (N = 6–13 neurons, 3 platings/mouse, 3 mice/genotype; one-way ANOVA p values corrected for multiple comparisons using Tukey’s post hoc test; WT Veh vs WT sB4, p < 0.0001 (****); WT sB4 vs WT sB4 + DAPT, ****p < 0.0001; WT sB4 vs V321L Veh, ****p < 0.0001; WT sB4 vs V321L B4, ****p < 0.0001). All other comparisons are statistically not statistically significant. D, Cortical neurons from embryonic WT (dark blue) and V321L mice (light blue; E18.5) were cultured for DIV3 and were stimulated with soluble ErbB4 (sB4), PI3K inhibitor WM, γ-secretase inhibitor L-685,458 (L6), WM + B4, or L6 + B4. Neurons that underwent no drug treatments/sB4 stimulation are indicated as the control group (C). Neurons were fixed and axonal length was quantified. (Two-way ANOVA with Tukey’s post hoc correction, WT C vs WT B4, ****p = 0.0002; WT L6 vs WT L6 + B4, **p = 0.0047; V321L C vs V321L B4, ***p = 0.001; V321L WM vs V321L WM + B4, p = 0.1; V321L L6 vs V321L L6 + B4, *p = 0.03.) N = 20–37 neurons per genotype per condition. ns, not significant. E, Treatment and conditions same as in D. Quantification is for dendritic length. (two-way ANOVA w/ Tukey’s post hoc correction: WT C vs WT B4, **p = 0.002; WT WM vs WT WM + B4, ****p < 0.0001). N = 20–37 neurons per genotype per condition. ns, not significant.
Type III Nrg1 back signaling results in the appearance of distinct clusters of the ICD in the nucleus (Bao et al., 2003). To determine whether nuclear ICD clusters were altered in neurons from V321L mice in response to ErbB4, we cultured dispersed hippocampal neurons from P4 WT and V321L mice [culture age, 17 d in vitro (DIV)]. We stimulated these cultures with soluble recombinant ectodomain of ErbB4 (sB4) and quantified nuclear ICD clusters (Fig. 2C, left). We found that stimulation with sB4 increased the number of nuclear ICD clusters in neurons from WT mice (Fig. 2C, right; WT Veh vs WT sB4 one-way ANOVA Bonferroni adj. p < 0.0001). This increase in nuclear ICD clusters was blocked by pretreatment with the γ-secretase inhibitor, DAPT [Fig. 2C, right; WT Veh vs WT (sB4) + DAPT Bonferroni adj. p > 0.9999]. We did not observe increased ICD clusters in neurons from V321L mice treated with sB4 [Fig. 2C, right; V321L Veh vs V321L (sB4) Bonferroni adj. p > 0.9999].
As mentioned before, Type III Nrg1 back signaling operates via two known mechanisms: (1) local axonal, PI3K-Akt signaling and (2) γ-secretase–dependent nuclear signaling, required for dendritic growth and complexity (Chen et al., 2010; Fazzari et al., 2014). We asked whether neurons from V321L mice are selectively deficient in dendrite development in response to ErbB4 stimulation. Cortical neurons from E18.5 WT and V321L mouse pups were cultured for DIV3. On the third day, neurons were treated with sB4 with or without pharmacological inhibition of PI3K using wortmannin (WM) or γ-secretase using L-685,458 (L6).
Neurons from WT mice showed increases in axonal and dendritic length in response to ErbB4 treatment which were blocked by WM and L6, respectively (Fig. 2D,E, dark blue boxes; axonal length, WT control vs WT sB4, p = 0.0002; dendritic length, WT control vs WT sB4, p = 0.0019). WM treatment did not block ErbB4-induced dendritic growth, and L6 treatment did not prevent sErbB4-induced axonal growth (axonal length, WT L6 vs WT L6 + sB4, p < 0.0001; dendritic length, WT WM vs WT WM + sB4, p < 0.0001). These results agree with previously published data showing that Nrg1 nuclear back signaling influences dendritic growth (Chen et al., 2010). These data also establish that stimulation of axonal growth requires PI3K signaling and that these two modes of Nrg1 back signaling are functionally independent.
Neurons from V321L mice showed increased axonal length in response to sErbB4 stimulation which was blocked by WM treatment and not by L6 (Fig. 2D, light blue boxes; axonal length, V321L control vs V321L sB4, p = 0.0014; V321L WM v. V321L WM + sB4, p = 0.1113; V321L L6 v. V321L L6 + sB4, p = 0.0289). On the other hand, V321L neurons did not show increases in dendritic length in response to sErbB4 stimulation (Fig. 2E dendrite length, V321L control v. V321L sB4 p > 0.9999). These results indicated that PI3K signaling-dependent axonal growth was intact in V321L mutant mice with a selective disruption of γ-secretase–dependent dendritic growth.
Thus, V321L mice show disruptions to regulated intramembrane proteolysis of Nrg1 and thereby to nuclear translocation of the ICD. Additionally, we demonstrate that dendritic growth in response to sErbB4 stimulation was absent in neurons from V321L mutant mice, whereas axonal growth in response to sErbB4 was intact.
Developmental regulation of Nrg1 nuclear back signaling in GC culturesIn hippocampal neurons, the Type III Nrg1 protein is part of the presynaptic membrane where it is predicted to interact with ErbB4 on dendrites of GABAergic interneurons (Vullhorst et al., 2009, 2017). We noted the presence of nuclear Nrg1 ICD clusters at the baseline in our previous experiments, which increased in number following stimulation with ErbB4 (Fig. 2C). Thus, we next sought to examine this baseline endogenous nuclear signaling and asked whether it might correspond to a specific developmental window. We first characterized our P4 hippocampal culture preparation to ask if GABAergic interneurons were present in our culture, as a possible source of ErbB4 in the culture. Approximately 60–70% (mean, 63% ± 10% SD) of the neurons were GCs (Prox1+), whereas ∼25% (mean, 25% ± 7.5% SD) of the neurons were GABAergic (GAD67+) and the remainder ∼12% (mean, 12% ± 12% SD) were other neurons likely corresponding to glutamatergic pyramidal and mossy cells (Fig. 3A). Next, we assessed baseline and stimulated nuclear back signaling at DIV10, 14, and 17. We found that the baseline nuclear back signaling diminished over time in culture but remained inducible by sErbB4 treatment (Fig. 3B; Kruskal–Wallis test, p = 0.0003; KW = 23.31; DIV10 control vs DIV17 control Dunn’s adj. p = 0.049; DIV17 control vs DIV17 + sB4 Dunn’s adj. p = 0.006; DIV10 + sB4 vs DIV17 + sB4 Dunn’s adj. p > 0.9999). Intriguingly, at DIV10 the level of baseline nuclear ICD clusters was high, and sErbB4 treatment could not significantly enhance this signal (Dunn’s adj. p > 0.9999). Since Nrg1 nuclear back signaling is thought to be regulated by cell–cell interactions and in turn influences neurite growth and development, we characterized the axonal and dendritic growth over time in our GC cultures to ask whether the high basal nuclear back signaling might correspond to a particular developmental process associated with neurite growth. Axonal growth (indicated by the area covered by SMI312+ processes) increased from DIV1 to DIV10, plateauing thereafter (Fig. 3C; Kruskal–Wallis, p = 0.0001; KW = 25.79; DIV1 vs DIV10 Dunn’s adj. p = 0.0003; DIV1 vs DIV14 Dunn’s adj. p < 0.0001; DIV5 vs DIV14 Dunn’s adj. p = 0.0462). Qualitatively, we noted a gradual increase in axonal bundling over time. Similarly, we noted a gradual increase in dendritic growth (indicated by the area covered by MAP2+ processes) from DIV1 to DIV14 (Fig. 3D; Kruskal–Wallis p > 0.0001; KW = 24.93; DIV1 vs DIV10 Dunn’s adj. p = 0.0035; DIV1 vs DIV14 Dunn’s adj. p < 0.0001; DIV5 vs DIV14 Dunn’s adj. p = 0.0308). Qualitatively, we also noted an enhancement of dendritic complexity between DIV10 and 14 (Fig. 3D). As noted earlier, nuclear back signaling was maximal at DIV10; thus we examined interactions between axons and dendrites at DIV5, 10, and 14 to assess whether an increase in axon–dendrite contacts corresponds to the high nuclear back signaling (Fig. 3E). We found that at DIV5, a few thin SMI312+ processes (individual axons) were in proximity to MAP2+ processes (dendrites; Fig. 3E, left). This was dramatically enhanced at DIV10 where we noted a higher amount of axonal coverage, along with multiple axons “running along” dendrites (Fig. 3E, middle). Finally, at DIV14, we noted axonal bundling indicated by the presence of thicker SMI312+ processes, which ran along dendrites (Fig. 3E, right). Thus, under these culture conditions, DIV10 represents a period of dynamic axonal growth, axon–dendrite contact, and endogenous nuclear back signaling, which is followed by increased dendritic complexity. Intriguingly, this period falls squarely within the γ-secretase inhibition–sensitive window for dendrite development (see below) indicating that the nuclear ICD might regulate genes related to dendrite development.
 Figure 3.
Figure 3. The peak of Nrg1 nuclear back signaling aligns with the peak of axon growth and precedes the peak of dendrite growth. A, Characterization of neuronal types present in the P4–5 hippocampal culture. Left, Representative image from a WT P5 culture stained for GAD67 (grayscale; GABAergic neuron marker), TuJ (green; neuronal marker), Prox1 (red; GC marker), and counterstained with DAPI (blue; to label nuclei). GCs were identified as Prox1+ TuJ+ cells. GABAergic interneurons were identified as GAD67+ TuJ+ Prox1− cells, where the GAD67 staining also delineated the neurites (example indicated by yellow arrowheads). Neurons not stained by Prox1 or GAD67 were identified as other excitatory neurons likely representing pyramidal and/or mossy cells. Scale bar, 10 µm. Right, Quantification of neuronal types as proportion of all neurons. Data were pooled from cultures imaged on various days in vitro (DIV5, 10, 14). Overall, GCs make up majority of the neurons in these cultures (mean, 62.6%; SD, 10.5%). N = 15–16 platings from 12 to 16 mice from two litters. B, Quantification of Nrg1 ICD clusters in nuclei of WT neurons from P4 to 5 hippocampal cultures at different DIVs under basal conditions (control) and after stimulation with sB4 (20 nM). N = 6–15 neurons/condition from platings made from three mice; Kruskal–Wallis test, **p = 0.005; KW = 10.59. (Dunn’s corrected multiple comparisons) DIV10 versus DIV17 p = 0.0047. Note: DIV17 group same as Figure 2C for comparison. C, Left, Representative thresholded images of SMI-312 staining (pan-axonal neurofilament marker) from WT mice at DIV1, 5, 10, and 14. Scale bar (red), 10 µm. Right, Quantification of fraction of area covered by SMI-312 staining normalized to the number of neurons in the imaging field. In WT cultures overall axonal coverage increased until DIV10 stabilizing thereafter. Kruskal–Wallis test, p < 0.0001 KW = 25.79. (Dunn’s corrected multiple comparisons) DIV1 versus DIV10, ***p = 0.0003; DIV1 versus DIV14, ****p < 0.0001; DIV5 versus DIV14, *p = 0.046. All other comparisons were not significant. N = 8–10 platings from 6 to 8 mice from two litters for each time point. D, Left, Representative thresholded images of MAP2 staining (dendrite marker at time points after DIV1) from WT mice at DIV1, 5, 10, and 14. Scale bar (red), 10 µm. Right, Quantification of fraction of area covered by MAP2 staining normalized to the number of neurons in the imaging field. Overall dendrite coverage increased till DIV14 in WT cultures. Kruskal–Wallis test, p < 0.0001; KW = 24.93. (Dunn’s corrected multiple comparisons) DIV1 versus DIV10, **p = 0.004; DIV1 versus DIV14, ****p < 0.0001, DIV5 versus DIV14, *p = 0.03. All other comparisons were not significant. N = 8–10 platings from 6 to 8 mice from two litters per genotype for each time point. E, Representative images of MAP2 and SMI312 staining of WT cultures at DIV5, 10, and 14 showing increased axon–dendrite interactions at DIV10. Yellow arrowheads denote regions of axon–dendrite contacts. Scale bar, 10 µm.
γ-Secretase activity antagonistically regulates axon and dendrite developmentThe V321L mutation disrupts γ-secretase processing of Nrg1 thereby preventing nuclear translocation of the ICD (Fig. 2C). Additionally, nuclear translocation of the ICD is regulated during development in GC-enriched cultures (Fig. 3B). Thus, we next queried the role of γ-secretase activity in axon and dendrite development during specific developmental periods. We noted that the levels of Nrg1 ICD in the nucleus peak at or prior to DIV10 after which they decline (Fig. 3B). Additionally, it was after DIV10 that we noted the dramatic increase in dendritic complexity (Fig. 3D, left). Thus, we hypothesized that inhibiting γ-secretase during this period might influence dendrite development. We treated P4 WT hippocampal cultures with the γ-secretase inhibitor, DAPT, or vehicle (control) for varying durations of time—DIV1–14, DIV5–14, or DIV12–14 (Fig. 4A, left). Cultures were fixed and stained for the pan-axonal marker SMI312 and dendritic marker MAP2 on DIV14 to quantify axonal and dendritic coverage (Fig. 4A, right). Inhibiting γ-secretase prior to DIV12 resulted in severe dendritic growth defects (Fig. 4B, top; Fig. 4C, Kruskal–Wallis test, p = 0.0003; KW = 19.09; control vs DAPT1–14, p = 0.015; control vs DAPT5–14, p = 0.003; control vs DAPT12–14, p > 0.9999; DAPT5–14 vs DAPT12–14, p = 0.011). Starting γ-secretase inhibition after DIV5 was sufficient to induce the severe dendritic growth defect phenotype, indicating that there is a period of γ-secretase–dependent dendrite growth between DIV5 and 12. We also found that there was an increase in the number of primary neurites immunoreactive for the pan-axonal marker, but no significant differences in axonal coverage between the control and γ-secretase–treated groups (Fig. 4B, bottom; Fig. 4D. SMI312+ primary neurites, Kruskal–Wallis test, p = 0.01; KW = 11.13; C vs DAPT5–14 p = 0.04; Fig. 4E, axonal coverage, Kruskal–Wallis test, p = 0.21; KW = 4.487). These results indicate that γ-secretase activity between DIV5 and 12 is necessary to constrain axonal development and promote dendritic development.
 Figure 4.
Figure 4. γ-Secretase regulates axon and dendrite development. A, Schematic of DAPT treatment regimens that cultured neurons from WT mice (P4/5 hippocampus) were subjected to. Vehicle controls (C) received equal volume of DMSO diluted in culture media. The DAPT1–14 group received DAPT treatment from DIV1–14, the DAPT5–14 group was cultured for the first 5 d without DAPT but received DAPT from DIV5 onward, and the DIV12–14 group received DAPT from DIV12 onward. All platings were collected at DIV14, fixed and stained for MAP2 and SMI312. B, Representative images of (top) MAP2 and (bottom) SMI312 immunostaining for each of the treatment conditions described in panel A. Asterisk (*) denotes the soma of each neuron. Magenta and green arrow heads indicate MAP2+ and SMI312+ neurites, respectively. Green circles in the MAP2-stained images mark the point(s) of origin of SMI312+ neurites. The same neuron is shown in both top and bottom panels. Note that for the DAPT DIV1–14 images, the SMI312 panel was recentered to allow visualization of the axon (soma now in bottom right corner). Scale bar, 10 µm. C, Quantification of fraction of area covered by MAP2 staining normalized to the number of neurons in the imaging field. Inhibition of γ-secretase prior to DIV12 significantly reduced dendritic growth. Kruskal–Wallis test, p = 0.0003; KW = 19.09. C versus DAPT1–14 Dunn’s corrected, *p = 0.015; C versus DAPT5–14 Dunn’s corrected, **p = 0.003; DAPT5–14 versus DAPT12–14 Dunn’s corrected, *p = 0.011. N = 6–8 platings from 6 to 8 mice from two litters per genotype for each time point. D, Quantification of number of primary neurites immunoreactive for SMI312 after vehicle control treatment or treatment with DAPT for varying durations. Kruskal–Wallis test, p = 0.01; KW = 11.13. C versus DAPT5–14 Dunn’s corrected, *p = 0.04. All other comparisons were not statistically significant. N = 6–8 platings from 6 to 8 mice from two litters per genotype for each time point. E, Quantification of fraction of area covered by SMI312 staining normalized to the number of neurons in the imaging field. Inhibition of γ-secretase had no significant effect on axonal coverage. Kruskal–Wallis test, p = 0.21; KW = 4.487. N = 6–8 platings from 6–8 mice from two litters per genotype for each time point.
Changes in Nrg1 V321L mutant DG transcriptome point to aberrant neurogenesis, cell cycle dynamics, and dendrite developmentThe Nrg1 ICD has strong transactivation properties (Bao et al., 2003), and V321L mice have impaired nuclear translocation of the ICD (Fig. 2). Thus, we predicted that impaired nuclear signaling by the Nrg1 ICD would result in transcriptomic changes. We extracted RNA from microdissected DG from WT and V321L mice for RNA sequencing (Fig. 5A). DEG analysis revealed 1,312 significantly dysregulated genes (colored dots, ANCOVA corrected p ≤ 0.1 and linear FC of ±1.25) between WT and V321L mice among which were genes specifically important for DG GC specification and function such as Prox1, Calb1, and Synpr (Fig. 5B; Extended Data Table 5-1). To identify possible functional alterations that might result from transcriptional dysregulation in V321L mice, we used IPA to find cellular processes that might be affected by the significantly dysregulated genes. Several processes along the neurodevelopmental trajectory were predicted to be disrupted in the V321L DG, including cell proliferation, differentiation, and dendrite development (Extended Data Table 5-2). Disease annotations in IPA indicated that dysregulated genes in the V321L DG were enriched in genes associated with SCZ susceptibility, indicating that the V321L mutation alters the transcriptional landscape in ways that have been associated with the genetic architecture of SCZ (Extended Data Table 5-3). We also found that dysregulated genes in V321L mice were enriched in cancer-associated genes, consistent with predicted changes in regulation of cell proliferation (Extended Data Table 5-3).
Table 5-1Differentially expressed genes in V321L mutant DG compared to WT DG. Differential gene expression between V321L and WT DG adjusted for biological sex and batch effects along with various statistical measures from ANCOVA. Download Table 5-1, XLSX file.
 Figure 5.
Figure 5. The V321L substitution in Nrg1 alters gene expression in the DG. A, Schematic and representative image of DG dissection from hippocampal slices; Mo, molecular layer; GCL, granule cell layer; Hi, hilus. B, Volcano plot for DEGs between V321L and WT DG. Each datapoint represents a single gene. Colored points delineate FC of at least 1.25 (blue dots, downregulated; red dots, upregulated in the mutant samples) with an adjusted p value of <0.1, and dark-colored points represent those with ANCOVA adjusted p value of <0.05. (N = 6 mice/genotype, 3 males/3 females). Prox1, Calb1, Synpr, Ezh2, Cit, and Chrm5 were chosen for validation via RNAscope in situ hybridization. The complete list of DEGs, statistics, and pathway analyses can be found in Extended Data Tables 5-1–5-3. C, Representative images of WT and V321L sections showing DAPI-stained DG GCL and staining for mRNA of the indicated genes in panel B. D, Quantification of RNAscope signal shown in panel A. [From top to bottom, WT vs V321L Welch’s t test (two-tailed)] Calb1, *p = 0.049; t = 2.679; df = 4.435; Synpr, *p = 0.04; t = 2.672; df = 5.992; Prox1, *p = 0.02; t = 3.174; df = 5.882; Ezh2, *p = 0.03; t = 3.031; df = 5.034; Cit, **p = 0.005; t = 4.352; df = 5.983; Chrm5, p = 0.0548; t = 2.740; df = 3.808. N = 4 mice/genotype.
Table 5-2Ingenuity pathway analysis (IPA) for functional and pathway annotations. (Top) Functional annotations of differentially expressed genes in the V321L DG compared to WT DG. (Bottom) Canonical pathways implicated by the differentially expressed genes in the V321L DG. P-values and predicted activation state and z-scores. Download
Comments (0)