
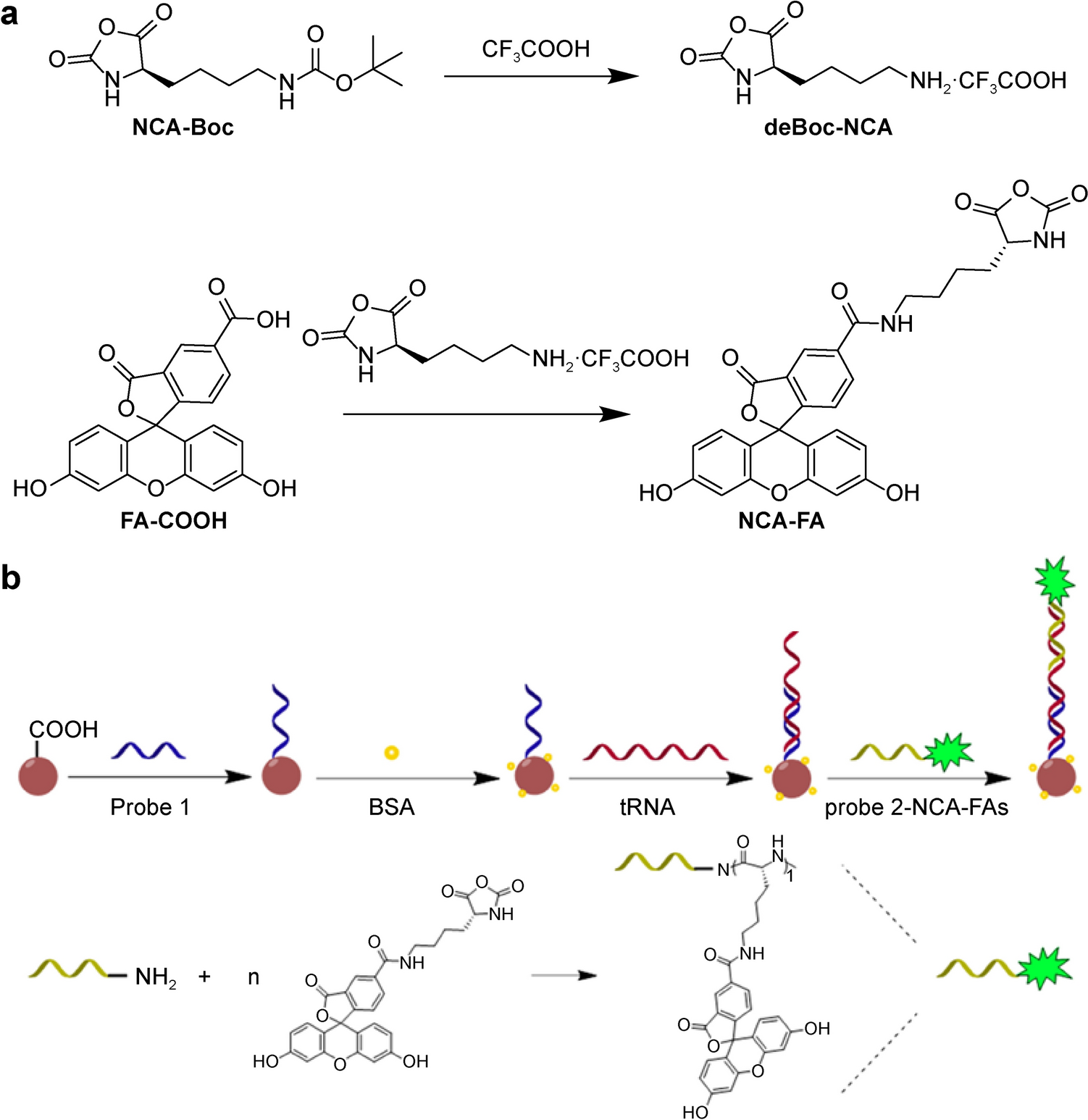
Remember me





Several chromatographic conditions regarding mobile phase composition, excitation and emission wavelengths, and column temperature, were tested to acquire and separate the signal of each analyte. Then, the compounds were singly injected to acknowledge their retention time.
Since this method was applied to biological samples, hence requiring analyte extraction and spiking with a predefined concentration of an IS avoid possible sample preparation errors and guarantee a reproducible and accurate quantification. Potential intrinsic fluorescent compounds, such as norfloxacin, ofloxacin, prulifloxacin, gatifloxacin, pazufloxacin, clinofloxacin, lacosamide, levetiracetam, zonisamide, perampanel, amitriptyline, antipyrine, pitavastatin, atorvastatin and 4-(Aminomethyl)benzene-1,2-diol were tested as IS. The last one was selected because it was the only one with a measurable peak, while none of the others were detected.
The mobile phase was composed of acetate buffer supplemented with EDTA and octane sulfonic acid. While this buffer shows good compatibility with fluorescence detection, EDTA improved peak shapes, and octane sulfonic acid, an ion-pairing reagent, increased the retention time of the compounds.
The pH of the acetate buffer was readjusted to 4.40 with acetic acid because it allowed the complete separation and resolution of the peaks. It is noteworthy that slight variations in pH strongly influence the resolution of DA, DOPAC, and HVA, as specified in Fig. 3.
Fig. 3
Influence of the mobile phase pH in peak resolution for each compound. The COOH/COO− group of DOPAC (pKa = 4.53) and HVA (pKa = 4.35) leads to variations in retention time and, consequently, peak resolution. TRYP also has a COOH/COO− group. However, TRYP is ionized at pH 4.30 and 4.40 (pKa = 2.36). The remaining compounds do not undergo variations at this pH because their NH2/NH3+ group grants a pKa > 8 (all non-ionized). The variation in DA resolution is related to the variation in retention time of HVA
Preparation and pretreatment of brain and PFC tissue samples were optimized to attain the best compound recuperation without interferences, initiated by tissue homogenization with ice-cold perchloric acid 0.2 mol/L in the proportion of 4 mL per g tissue. Perchloric acid solubilizes the compounds allowing their remotion from the tissue. Solid phase extraction was tested on Oasis HLB 1 mL 30 mg extraction cartridges (Waters™), however, the recoveries of compounds were meager (< 5%) with changes in the retention time. Liquid–liquid extraction (LLE) involves solvent evaporation under high temperatures. Since the neurotransmitters are temperature-sensitive, requiring sample manipulation on ice, LLE was not herein feasible. Therefore, PP extraction was optimized by testing different solvents. To 100 µL of brain homogenized, perchloric acid (0.2 mol/L, 1:2, V/V), perchloric acid (0.7 mol/L, 1:1, V/V), and acetonitrile (1:1, V/V) were tested resulting in a high sample dilution with consequent lower recoveries. To avoid sample dilution, perchloric acid (2 mol/L, 1:0.2, V/V) and trichloroacetic acid (20%, V/V) (1:0.2, V/V) were tested with recoveries ranging from 60 to 73%. The highest and most precise recuperation procedure was achieved with perchloric acid at 2 mol/L (1:0.1, V/V), with recovery values equal to or exceed 67%. Distinct times of vortex and centrifugation conditions, such as acceleration and time, were also investigated. Higher vortex and centrifugation time did not increase the recovery percentage leading to the final conditions, which are described in Sect. 2.4. However, under these experimental conditions, the stability of 5-HT was compromised, requiring the addition of cysteine to the ice-cold perchloric acid (0.2 mol/L) during homogenization. Different concentrations of cysteine (3, 6, and 9 mmol/L) were tested, and 6 mmol/L was selected as it ensured higher stability.
An isocratic elution, where the same proportion of acetate buffer and methanol is constant during all the running time, was the first approach. Moreover, there was an endogenous interferent at the retention time of NA, which demanded to optimize the chromatographic conditions. To a complete separation of the interferent from NA, it was necessary to decrease de proportion of methanol in the mobile phase. Nevertheless, TRYP, HVA, and 5-HT were eluted at higher retention times and with a broad peak shape. The complete separation was achieved recurring to a gradient elution as in Sect. 2.5.
The oven temperature was defined at 27 °C since it was the optimal condition for the most symmetric peak and best resolution without compromising the compounds stability.
The wavelengths for excitation (279 nm) and emission (320 nm) were defined based on the fluorescence spectrum of each compound and a diluted homogenized brain sample.
3.2 Full Method Validation in BrainFor total method validation in brain tissue, ICH Guideline M10 to Bioanalytical methods validation requirements must be fulfilled, namely selectivity, carry-over, specificity, calibration curve range and linearity, accuracy and precision, recovery, and stability [30].
Selectivity was evaluated by running six blank samples from independent donors, obtaining similar chromatograms. Nonetheless, neurotransmitters are endogenous compounds constitutively present in the brain, existing in the blank samples. According to the ICH Guideline M10 for Bioanalytical methods validation, since the interference of the endogenous compounds does not avoid an adequate signal-to-noise ratio, the background subtraction approach should be chosen for method validation [30]. This approach comprises pooling and mixing different brain homogenates to obtain a representative matrix, which will be used to construct the calibration curve. Blank samples (pool) are analyzed, and the endogenous concentrations measured will be subtracted from the areas observed on the calibration standard solutions. The obtained difference regards the concentration of the sample. Thus, the calibration curve was constructed resorting to the difference between the spiked matrix sample and the blank sample. The background subtraction approach allows an equal matrix effect because the study samples matrix is the same biological matrix used to construct the calibration curve [30].
An additional step to confirm selectivity was implemented using a double detection system encompassing DAD and FLD to compare the chromatographic runs of mobile phase spiked with one calibration standard, blank samples, and blank samples spiked with the same calibration standard (Fig. 4). The retention time of the compound was the same. The areas of the blank spiked sample would be the sum of the areas of the spiked mobile phase with the blank sample. Furthermore, overlapping the spiked mobile phase with the blank sample and the spiked sample confirmed the addition of areas without other interferences, supporting selectivity and peak purity (Fig. 4).
Fig. 4
Chromatogram of a a mobile phase spiked with the calibration standard P6, b a blank sample, and c a blank sample spiked with the fortification solution P6. IS (0.5 µg/mL) presents a retention time of 7.7 min and it is present in all spiked samples
The carry-over effect was also analyzed by running the highest QC sample. No peaks were observed in successive analytical runs at the retention times of the analytes.
Specificity was also performed particularly bearing in mind the in vivo studies herein were developed with cannabinoids (Sect. 2.7). Indeed, cannabinoids emit fluorescence and could interfere with the analytes under investigation [32]. We demonstrated that CBG, CBC, CBN, CBD, and CBDV did not interfere with the neurotransmitters, their metabolites, or precursors since their elution occurs with the mobile phase at void time.
Calibration curves and QC samples were defined according to the endogenous levels quantified in blank samples of brain tissue. During full validation, five calibration curves (n = 5) were independently tested for each neurotransmitter in brain-homogenized tissue. The calibration curves ranges were defined as follows: NA (10–400 ng/mL); AD (10–200 ng/mL); DA (10–800 ng/mL); DOPAC (50–300 ng/mL); HVA (55–300 ng/mL), TRYP (35–1000 ng/mL), and 5-HT (35–300 ng/mL). Since heteroscedasticity was confirmed for all the analytes except DOPAC, several weighting factors were tested and 1/x2 was selected as it ensured the smallest sum of absolute percentage relative errors for intra-day and the best weighted linear regression factor (r). The calibration parameters attained for each compound, including slope, interception, and regression coefficient (r2) are shown in Table 1. Good linearity was found for all analytes (r2 ≥ 0.988).
Table 1 Calibration curve parameters (n = 5) for NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in mice brain homogenateTo perform accuracy and precision, inter and intra-day tests were conducted using the QCLLOQ, QC1, QC2, and QC3. Precision and accuracy were recorded during five independent days (n = 5) for total validation, intra-day analysis was performed by the repetition of each QC five times on the same day (n = 5), while inter-day analysis was performed comparing each QC on five different days. The results are presented in Table 2, and all of them are within the required ranges admitted by the ICH Guideline M10 on Bioanalytical Method Validation defined, i.e. ± 20% in the QCLLOQ and ± 15% in the QC1, QC2, and QC3 [30]. Recovery of each neurotransmitter was investigated recurring to QC1, QC2, and QC3 by comparing the values obtained in the spiked samples subjected to the extraction process with spiked samples non-subjected to the extraction process for each compound. The recoveries of each neurotransmitter are presented in Table 3. The CV values were lower than 13.87%, demonstrating the precision of the sample preparation procedure.
Table 2 Inter and intra-day results of the mean experimental concentration (ng/mL) and respective accuracy and precision (n = 5) for NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in total mice brainTable 3 Relative recoveries of the NA, AD, DA, DOPAC, HVA, TRYP, and 5-HTThe absolute recovery of the IS (500 ng/mL) was (108.01 \(\pm\) 2.72) %, expressed as (mean \(\pm\) SD) %, which also proves precision in the sample preparation process.
Stability conditions were defined based on the sample preparation, storage, and the time to their analyses. The stability of unprocessed brain samples revealed that all the compounds were stable at 4 °C for 2 h (89.30%—109.47%, which is in the defined range by the ICH Guideline). Regarding processed samples, all compounds were stable for 24 h at 4 °C and 6 h at RT (the stability percentage was between 86.89% and 111.67% and in accordance with ICH). NA, AD, DA, and HVA were stable for 9 h at RT (the stability percentage was between 91.65% and 103.90%) (Table 4).
Table 4 Stability of NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in unprocessed and processed samplesSince the monoamines are endogenous compounds, according to the ICH Guideline, stability experiments should mimic as much as possible the study samples and should be analyzed in the authentic biological matrix [30]. In this way, the endogenous stability in the brain homogenates and brain tissue was also investigated, without spiking with any QC standard solution. Hence, at −20 °C for 3 d, only DOPAC and 5-HT were not stable in brain homogenate, while the remaining compounds were within 93.89% to 99.74%. All compounds were stable after one cycle of freezing and thawing of the brain tissue (94.1–111.74%, Table 5). Note that, the endogenous stability of AD was not possible to determine since the endogenous concentration was inferior to the LLOQ.
Table 5 Stability of NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in authentic biological matrix.The stability of the combined calibration standards and SS was demonstrated during 7 d at 4 °C (Table 6) as well as the IS standard solution (102.44%). However, the stability of the IS and SS was guaranteed during 7 d at −20 °C (100.43%).
Table 6 Stability of NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in calibration standard solutions3.3 Partial Method Validation in PFCEstablished the best experimental conditions and once validated, the HPLC method was partially validated in mice PFC matrix to quantify the analytes in vivo preclinical studies. According to ICH guideline, partial validation of the method ideally requires selectivity, carry-over effect, calibration curve range, linearity, as well as inter-day accuracy and precision [30].
Herein, selectivity was evaluated by running 6 different PFC homogenates obtained from independent animals. The chromatograms of 6 blank PFC homogenates were very similar to those observed for the total brain homogenates, revealing no additional peaks or statistical differences at the areas of the identified peaks. The carry-over effect was absent and the interferents was not observed from previous runs.
Considering the similarity between PFC and total brain homogenates, the calibration curve range applied in the PFC matrix was the same as the total brain matrix. Linearity was investigated through the construction of three calibration curves on independent days (n = 3) and a good linearity was observed for all analytes (r2 ≥ 0.986). The calibration parameters collected for each compound, including slope, interception, and regression coefficient (r2) are shown in Table 7.
Table 7 Calibration curve parameters of NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in mice PFC homogenateInter-day accuracy and precision were achieved recurring to the QCLLOQ, QC1, QC2, and QC3 and were recorded during three independent days (n = 3). The results are presented in Table 8, and all of them are within the required ranges admitted by the ICH Guideline M10 on Bioanalytical Method Validation defined, i.e. ± 20% in the QCLLOQ and ± 15% in the QC1, QC2, and QC3 [30]. The partial validation in PFC homogenates demonstrated reproducibility and can be applied in in vivo preclinical studies.
Table 8 Inter-day results of the mean experimental concentration (ng/mL) and respective accuracy and precision (n = 3) for NA, AD, DA, DOPAC, HVA, TRYP and 5-HT in PFC3.4 Method ApplicationTo investigate the impact of CBD, CBG, and CBDV on monoaminergic pathways, healthy male mice received one single dose of CBD, CBG, or CBDV (55 µmol/kg, ip, n = 4). The control group of healthy CD-1 male mice (n = 5) was administered with the vehicle. Animals were sacrificed 30 min post-administration to quantify the neurotransmitters, metabolites, and precursors at the PFC (Fig. 5).
Fig. 5
Representative chromatogram of a PFC sample collected from mice treated with 55 µmol/kg of CBD intraperitoneally
Except for AD, all neurotransmitters, their metabolites, and precursors were quantified in PFC matrices and were within the calibration curve ranges herein defined. In fact, AD has been also reported as unmeasurable with other analytical methods. For instance, Benedetto et al. stated AD concentrations were lower than LLOQ (6.0 ng/mL) in healthy mice [3]. Importantly, non-treated animals evidenced concentration ranges of NA, 5-HT, and DA in PFC similar to those previously reported in the literatures [3, 18, 24, 33]. Only two methods currently quantify neurotransmitters by HPLC-FLD without derivatization, however, they require higher injection volumes [3, 18]. Benedetto et al. only quantified four neurotransmitters (NA, DA, DOPAC, and 5- HT) in mice brain homogenates, with higher LLOQ for NA and DA, whereas the LLOQ of 5-HT was very similar to that reported herein (31 ng/mL vs. 35 ng/mL) [3]. The technique of Fonseca et al. also quantified DA, NA, AD and HVA but lacked neurotransmitters from the serotoninergic pathway. Moreover, it was developed in rat matrices and required gradient elution using two different buffers [18].
The levels of NA in the PFC of healthy mice 30 min following the administration of the cannabinoids were lower than those observed in the control group. Specifically, animals treated with CBDV presented statistical differences (p < 0.05, Fig. 6a) relative to the control group. In opposition, no statistical differences were found between the 5-HT concentrations in the PFC of treated and untreated animal groups. However, animals treated with CBD or CBDV presented 5-HT concentrations lower than those observed in CBG-treated animals (p < 0.05, Fig. 6b), suggesting that CBD and CBDV modulate the serotoninergic pathway in healthy animals. In addition, cannabinoids revealed a tendency to increase TRYP levels, particularly CBG (p < 0.05 compared to the control group, Fig. 6c), suggesting the potential of CBG to restore TRYP levels in CNS diseases in which it is decreased, such as major depressive disorder [34, 35] and bipolar disorder [36]. These results are innovative and only Jenny, M. et al. have already demonstrated that CBD modulates serotoninergic signaling increasing the availability of TRYP in mitogen-stimulated peripheral blood mononuclear cells [37].
Fig. 6
Results of neurotransmitters quantification. Concentrations of a noradrenaline, b serotonin, c tryptophan, d dopamine and dopamine metabolites, e DOPAC and f HVA in PFC 30 min after an acute intraperitoneal administration of CBD, CBG or CBDV (55 µmol/kg) to mice (n = 4). Results are represented as mean ± Standard Error of Mean (SEM). *p < 0.05 and **p < 0.01 as extracted from Kruskal–Wallis test, with multiple comparison Uncorrected Dunn’s test
CBD and CBDV also significantly decreased the levels of DA in PFC compared to the control group and the animals treated with CBG (p < 0.05, Fig. 6d). Complementarily, DOPAC and HVA, which are the main metabolites of DA, are enhanced after cannabinoid administration (Fig. 6e and f), even though statistical differences were found only between CBD and the control group for DOPAC. These findings suggest that CBD and CBDV can increase the metabolism of DA in PFC. In rats, Rossignoli, M. T. et al. reported a decrease in the DOPAC/DA ratio 5 d after intra-PFC microinjection of CBD [11].
These results evidence that this bioanalytical method could be accurately and precisely applied to demonstrate that cannabinoids modulate the monoaminergic pathways in healthy mice with a single intraperitoneal administration. Since this method has revealed its translational potential from brain tissue to PFC, it is expected to also be well succeeded if applied in other brain regions and after administration of other drugs or xenobiotics besides cannabinoids.
Comments (0)