
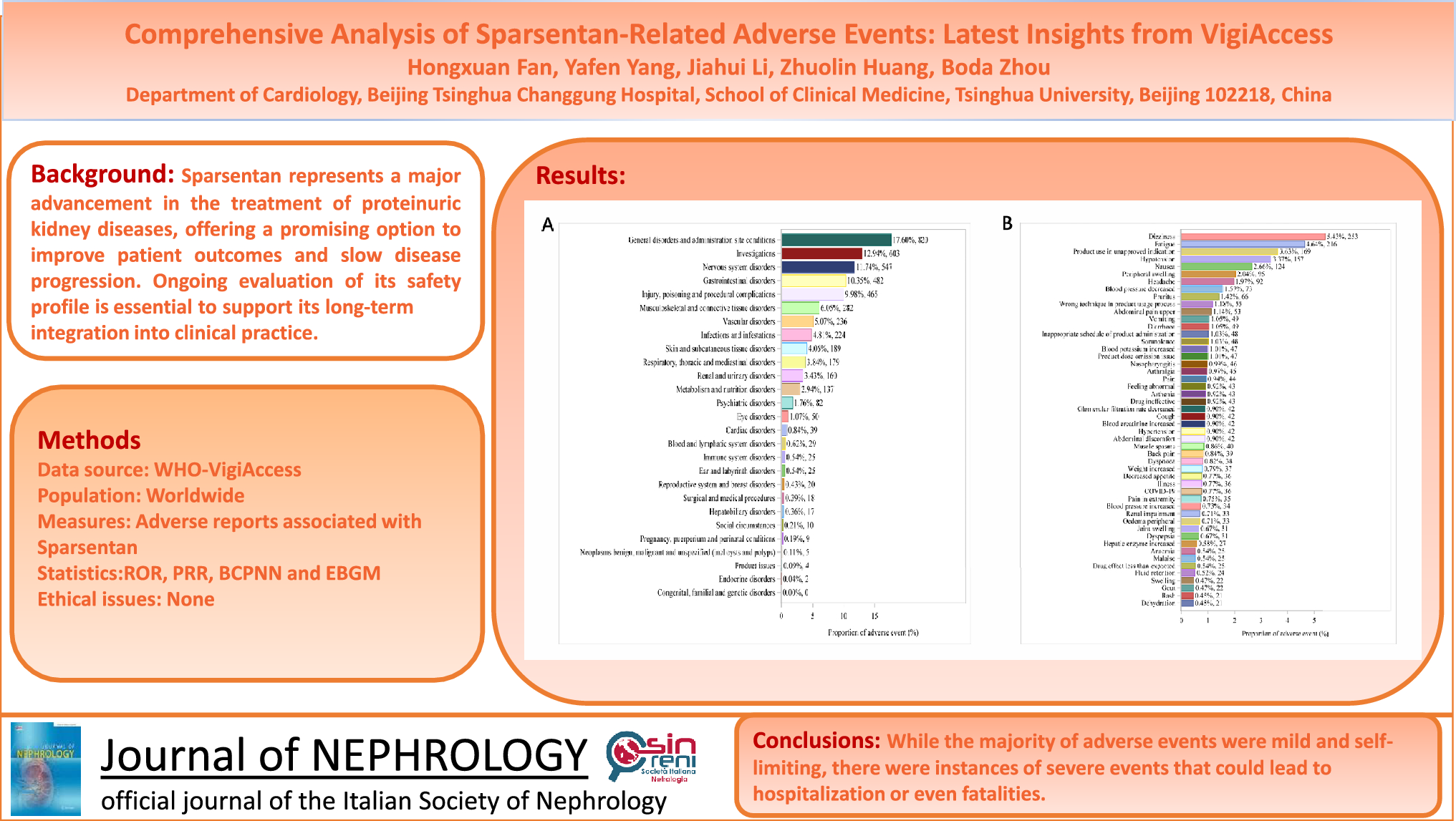
Remember me





Nephrotic syndrome is a rare complication of bone marrow transplantation but should always be considered, especially when peripheral oedema increases and kidney function deteriorates. Nephrotic syndrome is typically diagnosed approximately 6–12 months after alloHSCT. The incidence in adults is estimated to range from 0.37 to 6.1% [3]. In patients undergoing allogeneic bone marrow transplantation, the most common pathologies causing nephrotic syndrome are membranous nephropathy (MN) and minimal change disease (MCD) [2]. To confirm nephropathy, a kidney biopsy is usually necessary. In a study by Luo et al. [3], nephrotic syndrome was found in 9 of 257 patients who underwent alloHSCT. The 5-year incidence was 3.5%, with average onset occurring 225 days after transplantation. Among patients treated for chronic GvHD, nephrotic syndrome was diagnosed in 7 patients more than 3 months after alloHSCT [3]. In a recent study by John et al. [4], kidney biopsy was performed on 19 allogenic and 5 autologous HSCT recipients out of 2930 patients who underwent HSCT at the Department of Nephrology and Haematology at Christian Medical College, Vellore, South India, between 2005 and 2020. Among allogenic recipients, two clinico pathological patterns were found: thrombotic microangiopathy (TMA, 12/19 [63%]) and nephrotic syndrome, 7/19 [37%]. Of the 7 patients with nephrotic syndrome, MN was observed in 4 (57%) and MCD in 3 (43%). These patients were primarily treated with steroids, while MMF was used as second-line therapy. The authors noted that kidney biopsy was usually performed in HSCT patients with a strong suspicion of renal GvHD, such as concomitant GvHD at other sites, renal dysfunction, or proteinuria occurring after the discontinuation of GvHD prophylaxis.
In another study involving 1198 children after HSCT for either malignant or non-malignant conditions, 25 children underwent kidney biopsy [5]. The main pathology findings were mesangial proliferative glomerulonephritis (MPGN) and FSGS. Only three children had nephrotic-range proteinuria. Four children in this cohort had kidney injury prior to HSCT and their renal pathology showed FSGS and tubulointerstitial nephritis. After alloHSCT, 13 of these 25 paediatric patients exhibited evidence of GvHD, including 9 with acute GvHD and 4 with chronic GvHD, mostly involving the intestine, skin, and/or liver. In the only case of FSGS following HSCT for severe aplastic anaemia, no acute or chronic GvHD was observed [6].
A published case report discussed FSGS as a complication of GvHD in a 54-year-old man with multiple myeloma undergoing HSCT from a human leukocyte antigen-identical sister [7]. In a more recent study, Yap et al. [8] analysed clinical and histopathological data from patients who developed de novo glomerular diseases after HSCT, with 31 of 2222 patients (1.4%) showing de novo glomerular diseases after a mean duration of 2.8 ± 2.7 years post-HSCT. Histopathological diagnoses included TMA (38.7%), MN (25.8%), MPGN (12.9%), MCD (9.7%), FSGS (9.7%), and membranoproliferative glomerulonephritis (3.2%). Beyar-Katz et al. [9] analysed 116 cases of nephrotic syndrome diagnosed post-HSCT performed between 1988 and 2015, and found that the onset of nephrotic syndrome was associated with acute or chronic GvHD in 87.2% of cases. MN was the most frequent pathology (65.5%), followed by MCD (19%).
Glomerular diseases are an important complication in patients undergoing HSCT, affecting approximately 1–2% of all HSCT recipients, reaching up to 700–1400 cases per year worldwide. GvHD is currently considered the dominant aetiological factor for nephrotic syndrome after HSCT [10]. There is likely a causal relationship between cytomegalovirus infection, exposure to radiations, and the occurrence of haemolytic uraemic syndrome. It should be emphasised that nephrotic syndrome may be one of the symptoms of chronic GvHD and is usually diagnosed after discontinuing or reducing the dose of immunosuppressive drugs [10]. The occurrence of nephrotic syndrome is often accompanied by the recurrence of chronic GvHD. Furthermore, in a recent study, Beshensky et al. [11] suggested that kidney dysfunction in the context of less severe chronic GvHD may be unrelated to chronic GvHD and, potentially, is a consequence of drug-related toxicities. As the development of CKD in HSCT recipients is often multifactorial, a kidney biopsy is required to identify the underlying disease aetiology and pathology [12]. However, data on the kidney biopsies of patients with proteinuria after HSCT are extremely limited. Currently, no guidelines exist for the treatment of nephrotic syndrome after HSCT, but systemic treatment with corticosteroids and CsA is most widely used. The decision to initiate immunosuppressive treatment should be based on the patient's risk factors, together with the severity of proteinuria and kidney function [13].
Early trials leading to ruxolitinib approval did not report nephrotoxicity; however, a recent trial in patients with steroid-refractory atypical GvHD reported that 18% of participants experienced elevated serum creatinine and 4% of patients experienced a severe adverse event due to acute kidney injury (AKI) [A Study of Ruxolitinib in Combination With Corticosteroids for the Treatment of Steroid-Refractory Acute Graft-Versus-Host Disease (REACH-1) [Internet]. 2016. https://clinicaltrials.gov/ct2/show/results/NCT02953678. Accessed June 27th, 2025]. Strohbehn et al. [14] identified and reviewed all patients with a prescription for ruxolitinib between 2012 and 2019 (N = 414); the most common indication for ruxolitinib was atypical GvHD (47%). They found that moderate-to-severe AKI occurred within 90 days of starting ruxolitinib in 30 (10%) patients, 25 (83%) of whom were being treated for acute GvHD. However, no biopsy was performed. Recently, Ai et al. [15] reported a case of a person who developed post-HSCT nephrotic syndrome and portal hypertensive ascites. A kidney biopsy revealed membranous nephropathy and renal thrombotic microangiopathy with glomerular immune deposits, suggesting antibody-mediated kidney injury. Treatment with ruxolitinib resulted in remission of both nephrotic syndrome and ascites, suggesting a role for cytokines in the pathogenesis. In our case, after the initial success following ruxolitinib introduction, worsening kidney function and an increase in proteinuria were observed. Thus, it appears that ruxolitinib was a kind of ‘rescue’ therapy, albeit initiated too late to improve patient outcome.
This case illustrates the importance of a timely kidney biopsy as a guide for determining the extent of kidney damage and aiding in the decision-making process regarding therapy. As proteinuria increased despite nephroprotection, together with a rise in serum creatinine, a kidney biopsy was proposed and performed and it became clear that nephroprotection and preparation for kidney replacement therapy remained the optimal strategies. In our case, the kidney biopsy was performed too late, although at the earliest possible time upon referral to our department. As the person was young and in complete remission from haematological disease, kidney transplantation was considered and discussed. Therefore, the results of kidney biopsy are of additional value in cases of graft glomerulopathy to differentiate between recurrent and de novo changes. We would also like to stress the importance of a multidisciplinary team approach with the use of various therapeutic agents, including ruxolitinib, to manage GvHD and nephrotic syndrome. We have also included a proposed flow chart with management suggestions in post HSCT nephrotic syndrome (Fig. 3).
Fig. 3
Flow chart with management suggestions in post HSCT nephrotic syndrome. HSCT haematopoietic stem cell transplantation, PLAR2R phospholipase 2 receptor, FAT1 FAT Atypical Cadherin 1(protoadherin), MN membranous nephropathy, CNI calcineurin inhibitors, MMF mycophenolate mofetil, FSGS- focal segmental glomerulosclerosis
Comments (0)