All melting points (m.p.) are uncorrected. HRMS were done on a X500D QTOF system (ABSciex) available at the CGS of the University of Pavia. 1H, 13C and 15N NMR spectra were recorded in DMSO-d6 on a Bruker AV NEO 700 MHz spectrometer, equipped with a triple resonance helium cooled cryoprobe. Chemical shifts are expressed in ppm from the solvent residual peak (δ) and coupling constants (J) are in Hertz (Hz): b, broad; s, singlet; bs, broad singlet; d, doublet; t, triplet; m, multiplet. IR spectra (nujol mulls) were recorded on a spectrophotometer available at the Department and absorptions (ν) are in cm−1. Chromatographic details are reported in the starting and reference material section reporting the corresponding relative retention times (r.r.t.).
Starting and reference materials
No authentic sample of the reported Prolylrapamycin 2 was available as a marker for its structure confirmation. As part of the study, the HPLC method used for the analysis of Rapamycin 1 was also used to evaluate the isolation and the purity of the peak at r.r.t 0.71, supposed to be the Prolylrapamycin 2. The analyses of Rapamycin 1 were conducted by analytical HPLC on a Thermo BDS Hypersil column C18, 4.6 × 100 mm, 3 µm, with a mobile phase consisting of acetonitrile/ammonium acetate buffer 50/50 (v/v). The Rapamycin 1 injection sample was eluted using an isocratic run. A flow rate of 1.0 ml min−1 and UV detection at 278 nm were used.
Structural elucidation of impurities in drug substances is per se an improvement, with favorable impact both on knowledge and on the state of control of pharmaceutical processes. In this specific case, the clearly achieved attribution of Prolylrapamycin structure definitely confirmed our previous understanding that this typical impurity of Sirolimus manufacturing process is not originated by degradation of the drug substance. It is a process-related impurity. More specifically, a fermentation byproduct. A final assessment about this feature is essential from regulatory perspective as well (e.g. ICH Q3, Classification of Impurities).
Prolylrapamycin 2 was achieved via isolation of a small quantity (ca. 10–15 mg) of that impurity from the fermentation broth in the plant. A sample from the fermentation plant was taken for the laboratory trials. Fermentation broth for Rapamycin 1 contained around 2% (Area % in HPLC analysis) of an impurity having r.r.t. 0.71. The sample was enriched in that impurity initially by treatment on silica gel flash chromatography with eluent the mixture EtOAc/n-heptane in gradient proportion from 7/3 to 9/1. In this way a sample containing around 15% (Area % in HPLC analysis) of the impurity at r.r.t. 0.71 has been obtained (ca 1.0 g).
A Shimadzu preparative HPLC equipped with LC-20AP pumps, CBM-20A System controller, SPD20A UV–Vis detector, FRC10A Fraction collector and SIL10AP injector was used for preparative isolation of the impurity Prolylrapamycin 2 (r.r.t. 0.71). The injector was equipped with 5.0 ml sample loop and used in manual injection. Symmetry Prep C18, 30 × 100 mm, 5 µm particle size (Waters Inc, USA) was employed for isolation of Prolylrapamycin 2. The flow rate was set at 20 ml min−1 and detection was carried out at 278 nm. Mobile phase consisting of A water and B acetonitrile was used. The following linear gradient was used: 0–1 min 50% B; 1–11 min increase from 50% B to 100% B; 11–20 min elution with 100% B; return to 50% B. Fractions containing Prolylrapamycin 2 were pooled and concentrated under reduced pressure on a Heidolph Rotavapor Model Laborota4000 to remove acetonitrile/water affording 20 mg of Prolylrapamycin 2.
Characterization of compound
1
A sample of Sirolimus 1 (6.6 mg, 7.22 mmol) was dissolved in 0.5 ml DMSO-d6. 1D and 2D NMR spectra were collected at a 700 MHz NMR instrument at 298 K. For a complete characterization of the structure of compound 1 FT-IR spectra were collected as well as HRMS spectra and DSC analysis gave indication of the meting point of the product and relative decomposition upon heating.
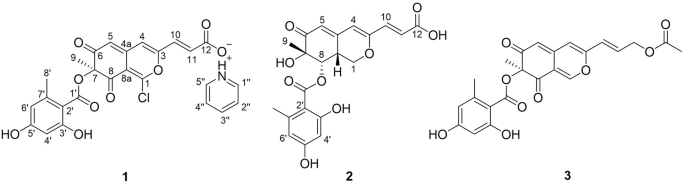
Compound 1: straw yellows crystals. DSC analysis: mp 189.68 °C followed by endo peak due to immediate decomposition below 202.80 °C. IR: νOH 3412, νC=O 1716, νC=C 1644 cm−1. 1H NMR (DMSO-d6) δ: 0.60 and 1.91 (m, 1H + 1H, CH2); 0.74 (d, 3H, J = 7 Hz, CH3); 0.78 (d, 3H, J = 7 Hz, CH3); 0.83 (d, 3H, J = 7 Hz, CH3); 0.87 (d, 3H, J = 7 Hz, CH3); 0.97 and 1.06 (m, 1H + 1H, CH2); 0.99 (d, 3H, J = 7 Hz, CH3); 1.04 and 1.41 (m, 1H + 1H, CH2); 1.25 (m, 1H, CH); 0.86 and 1.53 (m, 1H + 1H, CH2); 1.18 and 1.75 (m, 1H + 1H, CH2); 1.19 and 1.83 (m, 1H + 1H, CH2); 1.26 and 1.86 (m, 1H + 1H, CH2); 1.41 and 1.66 (m, 1H + 1H, CH2); 1.28 and 1.59 (m, 1H + 1H, CH2); 1.53 (m, 2H, CH2); 1.59 and 2.11 (m, 1H + 1H, CH2); 1.64 (s, 3H, CH3); 1.68 (m, 1H, CH); 1.75 (s, 3H, CH3); 2.04 (m, 1H, CH); 2.23 (m, 1H, CH); 2.37 and 2.74 (m, 1H + 1H, CH2); 2.42 (m, 1H, CH); 2.83 (m, 1H, CH); 3.06 (s, 3H, OCH3); 3.16 (s, 3H, OCH3); 3.18 (m, 1H, CH); 3.18 and 3.44 (m, 1H + 1H, CH2); 3.27 (m, 1H, CH); 3.33 (s, 3H, OCH3); 3.64 (dd, 1H, J = 12, 2 Hz, CH); 3.96 (d, 1H, J = 5 Hz, CH); 4.01 (m, 1H, CH); 4.02 (m, 1H, CH); 4.60 (m, 1H, OH); 4.94 (d, 1H, J = 7 Hz, CH); 4.99 (m, 1H, CH); 5.09 (d, 1H, J = 10 Hz, CH=); 5.27 (m, 1H, OH); 5.47 (dd, 1H, J = 15, 9 Hz, CH=); 6.12 (dd, 1H, J = 11, 2 Hz, CH=); 6.14 (dd, 1H, J = 16, 11 Hz, CH=); 6.23 (dd, 1H, J = 15, 11 Hz, CH=); 6.41 (s, 1H, CH=); 6.47 (s, 1H, OH). 13C NMR (DMSO-d6) δ: 10.96; 13.85; 13.94; 16.03; 16.08; 15.21; 20.86; 22.14; 24.95; 26.72; 26.92; 30.11; 31.61; 32.99; 33.38; 33.85; 35.29; 35.69; 35.93; 38.88; 39.98; 40.06; 40.32; 40.52; 43.99; 45.70; 51.25; 55.96; 57.25; 57.42; 66.50; 73.71; 74.02; 76.23; 82.72; 84.22; 85.98; 99.51; 125.39; 127.49; 127.54; 130.93; 132.81; 137.63; 138.35; 139.79; 167.48; 169.70; 199.39; 208.05; 210.99. HRMS for C51H79NO13: calcd. 913.5551. Found: 913.5511.
Characterization of compound
2
A sample of Prolylrapamycin 2 (3.2 mg, 3.55 mmol) was dissolved in 0.5 ml DMSO-d6. 1D and 2D NMR spectra were collected at a 700 MHz NMR instrument at 298 K. For a complete characterization of the structure of compound 2 FT-IR spectra were collected as well as HRMS spectra and DSC analysis gave indication of the meting point of the product and relative decomposition upon heating.
Compound 2: straw yellows crystals. DSC analysis: large band starting from 123 °C and ending after 200 °C, centered at mp 161.14 °C; decomposition follows the melting with change of colour of the sample. IR: νOH 3404, νC=O 1716, νC=C 1634 cm−1. 1H NMR (DMSO-d6) δ: 0.61 and 1.87 (m, 1H + 1H, CH2); 0.71 (d, 3H, J = 7 Hz, CH3); 0.77 (d, 3H, J = 7 Hz, CH3); 0.81 (d, 3H, J = 7 Hz, CH3); 0.83 (d, 3H, J = 7 Hz, CH3); 0.70 and 1.54 (m, 1H + 1H, CH2); 0.99 (d, 3H, J = 7 Hz, CH3); 1.00 and 1.40 (m, 1H + 1H, CH2); 1.24 (m, 1H, CH); 0.95 and 1.12 (m, 1H + 1H, CH2); 1.16 and 1.74 (m, 1H + 1H, CH2); 1.15 and 1.86 (m, 1H + 1H, CH2); 1.17 and 1.89 (m, 1H + 1H, CH2); 1.78 and 1.81 (m, 1H + 1H, CH2); 1.90 and 2.22 (m, 1H + 1H, CH2); 1.58 (m, 2H, CH2); 1.62 (s, 3H, CH3); 1.76 (m, 1H, CH); 1.78 (s, 3H, CH3); 2.05 (m, 1H, CH); 2.24 (m, 1H, CH); 2.25 (m, 1H, CH); 2.29 and 2.72 (m, 1H + 1H, CH2); 2.81 (m, 1H, CH); 3.07 (s, 3H, OCH3); 3.16 (m, 1H, CH); 3.18 (s, 3H, OCH3); 3.18 (m, 1H, CH); 3.23 (m, 1H, CH); 3.24 and 3.56 (m, 1H + 1H, CH2); 3.31 (s, 3H, OCH3); 3.67 (dd, 1H, J = 11, 2 Hz, CH); 4.03 (m, 1H, CH); 4.05 (m, 1H, CH); 4.18 (d, 1H, J = 3 Hz, CH); 4.23 (dd, 1H, J = 8, 4 Hz, CH); 4.59 (m, 1H, OH); 4.88 (m, 1H, CH); 5.12 (d, 1H, J = 10 Hz, CH=); 5.25 (m, 1H, OH); 5.47 (dd, 1H, J = 15, 10 Hz, CH = ); 6.14 (dd, 1H, J = 15, 11 Hz, CH=); 6.22 (d, 1H, J = 15 Hz, CH=); 6.29 (dd, 1H, J = 15, 11 Hz, CH=); 6.43 (dd, 1H, J = 15, 11 Hz, CH=); 6.50 (s, 1H, OH). 13C NMR (DMSO-d6) δ: 10.77; 12.71; 14.88; 15.43; 15.98; 16.11; 22.01; 24.54; 26.44; 29.40; 29.92; 31.37; 33.00; 33.06; 33.47; 34.69; 35.66; 35.95; 39.23; 39.79; 40.49; 40.85; 41.27; 44.98; 47.50; 55.98; 57.07; 57.24; 59.00; 66.34; 73.62; 73.75; 76.33; 82.43; 84.26; 85.16; 99.75; 124.43; 127.61; 127.71; 131.04; 132.95; 137.01; 138.21; 139.58; 166.03; 170.92; 198.66; 208.20; 210.03. HRMS for C50H77NO13: calcd. 899.5395. Found: 899.5336.


Comments (0)