
Remember me



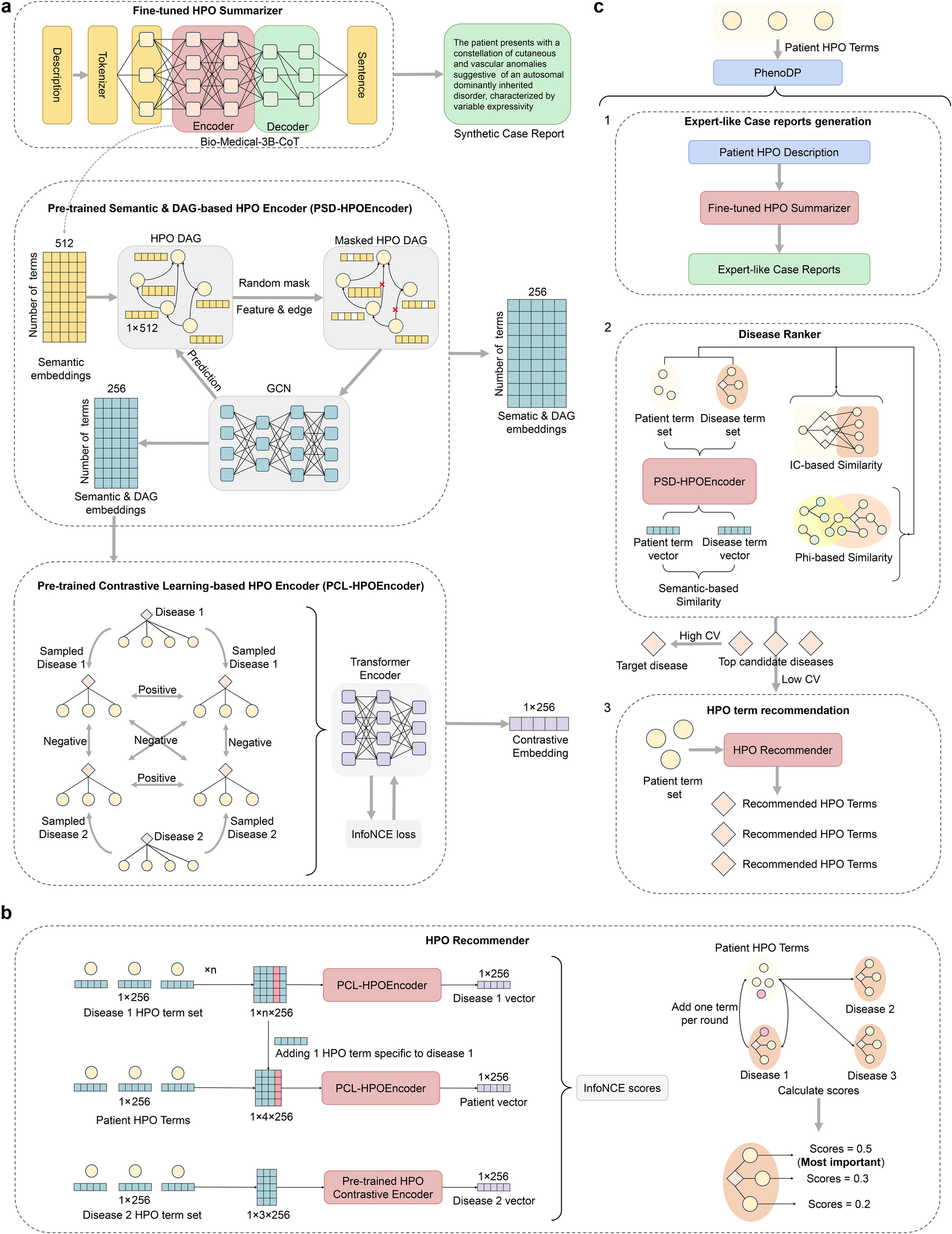



Implementation of viral metagenomic diagnostics and routine genomic surveillance can be particularly challenging due to the lack of computational infrastructure, tools, and/or bioinformatics expertise. In order to face the latter challenge, we have previously developed and openly released INSaFLU (https://insaflu.insa.pt/) [19, 22], a user-friendly bioinformatics suite for virus NGS data analysis. In the present study, we developed a new module (TELEVIR) for metagenomics virus identification, and considerably expanded and reinforced its genomic surveillance modules. Currently, INSaFLU-TELEVIR (https://insaflu.insa.pt/) [22] is an open web-based (but also locally installable; https://github.com/INSaFLU/docker) [68] bioinformatics platform for virus metagenomic detection and routine genomic surveillance that can be freely accessed upon account creation (user-restricted accounts). It can handle NGS data (single and/or paired-end data) obtained from different technologies (Illumina, Ion Torrent, and ONT), and derived from diverse wet-lab protocols (amplicon-based workflows, shotgun metagenomics, etc.) and library preparation/sequencing kits. It integrates two main analytical components: i) a virus detection pipeline: from NGS reads to quality control and metagenomics virus identification and reporting; ii) a reference-based genomic surveillance pipeline: from NGS reads to quality control, mutations detection, consensus generation, virus classification, alignments, “genotype-phenotype” screening, phylogenetics and integrative phylogeographical and temporal analysis, etc. (Fig. 1). An up-to-date documentation providing extensive usage example of data upload, analysis and management, and pipeline details (complementing the code available at https://github.com/INSaFLU) [71] and an extensive tutorial on how to upload data, run analysis, and visualize/download graphical and sequence/phylogenetic outputs is available at https://insaflu.readthedocs.io/en/latest/ [72], since its first release [19].
UsageFollowing the original web interface architecture [19], the upgraded INSaFLU-TELEVIR dashboard and functionalities are organized in four main interactive tabs: Settings, References, Samples, and Projects.
The Settings menu (a new feature since Borges et al., 2018) [19] is organized, when applicable, by module (Quality Control, INSaFLU, and TELEVIR), NGS technology, pipeline step, software, and parameters. This menu should be consulted to change specific software or controlling workflows, in order to fit the desired bioinformatics pipeline to the user’s needs, sample characteristics, and/or the upstream experimental conditions. For example, the default reads end’s trimming size may be too permissive or restrictive depending on the laboratory protocol (e.g., tiling amplicon multiplex PCR) and/or on sequencing settings or bioinformatics that were applied upstream (e.g., if reads are or not already trimmed/clipped before upload). The workflow and parameters selected in the global Settings menu are applied to the user account as a whole (i.e., to new samples and projects), but specific settings can be modified later on for individual samples or projects, in the respective menus. The References menu includes publicly available sequences (from NCBI or made available in INSaFLU under permission from the authors) to be used for reference-based genome assembly through “INSaFLU” projects (see below). It has been continuously enriched with sequences relevant for surveillance of viruses of interest, namely influenza, SARS-CoV-2, MPXV, and RSV. Similarly to the first platform version, additional reference files (FASTA and GenBank) can be uploaded to the user-restricted account. The Samples menu is the main sample repository, in which NGS reads (fastq.gz format), as well as the sample contextual data (i.e., metadata table in “csv” or “tsv” format, according to downloadable templates), are uploaded (through single upload or batch upload) or deleted. This menu also provides read-quality reports, technology identification, and rapid classification data (all automatically provided after upload), as previously described [19]. The main Projects menu allows access to three types of scalable projects: TELEVIR projects (for virus detection), INSaFLU projects, and Nextstrain datasets (both for virus routine genomic surveillance). The usage and functional and reporting features of these three main analytical modules are described below.
Metagenomics virus detectionTELEVIR projects—from reads to virus detectionOur benchmarking results consolidated the expectation that there is no “one-size-fits-all” bioinformatics approach that can detect all viruses, but instead a set of “well-performing” workflows that together can potentiate the detection of clinical relevant viruses, as described in the implementation section (and detailed Additional file 1 and Fig. 3). As such, the TELEVIR dashboard was designed to accommodate this flexibility by allowing users to simultaneously select complex workflows (covering several combinations of classification algorithms, databases, and parameters) in a user-friendly manner through the TELEVIR Settings pages. Controlling workflows is done by selecting/deselecting which software (and their parameters and/or databases, when permitted) are to run at each step of the pipeline (summarized in Fig. 2). Some key pipeline steps (e.g., confirmatory re-mapping) cannot be turned OFF. Other cases are context-dependent: de novo assembly cannot be turned OFF if Contig Classification is turned ON; at least one classification step must be turned ON (Contig Classification may not be turned OFF if Read Classification is already OFF, and vice-versa).
Fig. 3
Simplified illustration of the benchmark of the virus identification pipeline (TELEVIR) module components, which is described in detail in Additional file 1. A Tree representation of module combinations. From left to right, sections represent pipeline steps (exemplified for Illumina) as followed at runtime: (1) Quality Control, (2) Viral Enrichment, (3) Assembly, (4) Contig Classification, (5) Read Classification. Nodes represent software, parameters, or databases compared. Color gradient corresponds to the product of four assessment statistics: mapped reads proportion, horizontal coverage, true positive rate, and completeness (proportion of hits with both read and contig evidence). Statistics were standardized by their respective maxima. B Heatmap representation of software benchmarked for Illumina samples, parameters not discriminated, color code at the bottom. C Table of individual statistics for each node, standardized across samples as in A. For panels A and C, darker colors = lower values, lighter colors = higher values
In parallel, efforts were employed to develop and implement user-friendly (visual) solutions for output reporting. As the interpretation of metagenomics virus detection data is not a trivial task (even for users with expertise in virology and/or bioinformatics), the design of the TELEVIR output dashboard gave emphasis not only to increasing report accessibility and interpretation, but also to facilitating output navigation and promoting decision-making on the part of the users (especially relevant in clinical virology). Targeting these goals, TELEVIR reports are generated per workflow, per sample (combining several workflows), and per project (combining several samples), with a decreasing level of detail. Workflow reports are organized as dynamic and interactive “expand-and-collapse” panels that allow the visualization/download of relevant intermediate tabular (e.g., list of the software parameters, list of viral hits classified from reads and/or contigs) and sequence output data (e.g., reads surviving the viral enrichment and/or host depletion steps) generated throughout each workflow step (listed in Table S8; Fig. 4). Ultimately, each workflow culminates in a main report (interactive table) with a list of the detected top-viral hits, each one accompanied by several robust and diagnostic-oriented metrics, statistics, and visualizations (also detailed in https://insaflu.readthedocs.io/en/latest/) [72], provided as (interactive) tables, graphs (e.g., coverage plots, Integrative Genomics Viewer visualization, assembly to reference dotplot) and multiple downloadable output files (e.g., mapped reads/contigs identified per virus; reference sequences, BAM files, etc.) (Table S8; Fig. 4). In brief, the reported hits are identified (as detailed in the “Implementation” section), up to a user-defined maximum number of hits, as follows: reads and contigs (if available) are classified independently, then viral hits (TAXID) detected in both intermediate classification reports (reads and contigs) and/or within the top list from each side are selected for reference-based mapping against viral genome sequences present in the available databases. In summary, the main tabular report only includes viral hits (listed by the reference NCBI ACCID, with a direct interactive link to the NCBI webpage) that were classified at reads and/or contig level (“classification success”) and that had mapped reads or contigs (“mapping success”). Other viruses (TAXID) that were not automatically selected for confirmatory remapping are flagged as “Unmapped” and can be user-selected for mapping at any time through the bottom panel “Raw Classification and Mapping Summary” (which also lists hits yielding zero mapping). This functionality allows users to confirm/exclude the presence of a suspected virus (e.g., virus compatible with the animal/human clinical status) that did not meet the criteria for confirmatory remapping (e.g., due to their insufficient number of hits in the intermediate reports). Sample reports (interactive and downloadable tables) compile all viral hits identified in the main reports of the several workflows that were run for each sample, in which redundant hits are excluded (Fig. 4). Finally, Project results are provided as simple tables combining all top viral hits identified in the main reports of the several workflows that were run for all samples included in the project. Both Sample and Project reports provide direct links to the detailed reports generated at the workflow level for an enhanced sample comparison and output interpretation.
Fig. 4
Snapshot of dashboard reports of the INSaFLU-TELEVIR bioinformatics module for metagenomics virus detection. Interactive examples are available at the https://insaflu.insa.pt/ [22] through an open “demo” account
Besides the availability of multiple reports and downloadable files (summarized in Table S8), other important features were incorporated in the TELEVIR dashboard and documentation to facilitate the detection, evaluation and/or resolution of specific situations or confounding factors commonly faced during metagenomic NGS in clinical virology, in light of recent recommendations [18, 26, 27, 73].
Negative and positive controlsThe inclusion of negative controls (e.g., pathogen-negative samples, library preparation buffers) is highly recommended to identify sources of potential contamination and detect false positive hits [26, 27, 73,74,75,76,77]. Indeed, viral taxa/sequences detected in the test samples that are also present in the negative run controls should be interpreted as contamination or background noise (e.g., nucleic acids present in reagents might yield false positive viral hits across test and control samples). In addition, the inclusion of positive controls (e.g., samples spiked with viruses that cannot be found in the organism or in the environment that is being investigated) is also commonly performed to control the success of nucleic acids extraction, preparation, and sequencing [18, 26, 27, 73]. As such, TELEVIR users are encouraged to create different projects for different metagenomics sequencing runs, as they are allowed to select “control” sample(s) at any time (before and after data analysis) for each project. Viral TAXIDs detected in the main report of the user-selected “control” sample(s) are automatically flagged as “Taxid found in control” in the reports of samples in the same project. This functionality is designed to facilitate the background subtraction of viral hits also found in controls.
False-positive viral hitsIn the context of diagnostics, false-positive bioinformatics classification results can have significant consequences for patient/animal care [18, 26, 26, 73]. As such, TELEVIR reports provide specific warnings for two bioinformatics “artifacts” commonly yielding false-positive virus assignments: (i) “Vestigial Mapping” warning: when only a vestigial number of reads is mapped; (ii) “Likely False Positive” warning: when most read map in a very small region of the reference sequence, i.e., hits with high “DepthC” (mean depth of coverage exclusively in the covered regions) but low “Depth” (mean depth of coverage throughout the whole genome) and low “Cov (%)” (horizontal coverage) (specific flag criteria are detailed in Table S8). Of note, during benchmarking and testing, we noticed that both situations are often due to low-complexity regions (e.g., homopolymeric tracts or repeat regions). In this regard, an extra optional step of reads filtering by sequence complexity (using PrinSeq + +) [29] was added to the pre-processing step.
Multiple hits for several closely-related virusesCross-mapping of reads across several viruses (TAXID) with considerable nucleotide homology, such as viruses belonging to the same family, is very common in viral metagenomics. The interpretation of these cases is expected to be facilitated by the fact that the virus actually present in the sample is likely more closely related to the reference virus (TAXID) yielding the best TELEVIR mapping metrics (see Fig. S6), but extra manual inspection (namely, BLAST of mapped reads/contigs and IGV inspection) is recommended (see documentation and literature [26, 27, 73]. To further facilitate the report interpretation, viral hits included in the main reports (at both “workflow” and “sample” levels) are grouped and sorted by the degree of overlap of cross-mapped reads, as detailed in implementation. In addition, an optional and flexible step of “mapping stringency” is available to facilitate the detection of reads with high homology to the reference. Of note, by design, a true positive viral detection in TELEVIR will normally yield multiple hits for the same virus (TAXID). Two main situations justify this output: (i) the presence of segmented viruses in the sample (usually each reference segment has different ACCIDs, so they are reported as independent hits); (ii) the availability of several reference genomes (strains or variants) of the same virus in the databases. As above, in the latter situation, the virus present in the sample is likely more closely related to the reference genome (ACCID) yielding the best mapping metrics. The sorting strategy described above is expected to largely facilitate the report interpretation in these cases.
Although the INSaFLU-TELEVIR platform takes advantage of several viral reference databases, these do not cover all viruses. For instance, newly discovered or uncommon viruses or viral strains (e.g., viruses without available complete genomes in the databases) might be missing, leading to false negative results. Moreover, the ultimate goal of the TELEVIR module is to detect viruses (especially clinically relevant viruses), and not necessarily to identify the virus “strain/variant/serotype”. Once a given virus is detected, users are encouraged to perform fine-tuned analyses (e.g., consensus sequences reconstruction, mutation detection, etc.) using the classical INSaFLU projects (see below) to better characterize the virus found. Ultimately, in order to facilitate and strengthen the TELEVIR output interpretation and decision-making from the part of users, we highlight the availability of extended user guidance on how to interpret TELEVIR reports and exclude/confirm viral hits, by exemplifying “expected” metrics profiles (or combination of profiles) when there are different levels of evidence for the presence of a given virus in metagenomic NGS data analyzed through TELEVIR (https://insaflu.readthedocs.io/en/latest/) [72].
As described in the Implementation section, apart from the development and release of the TELEVIR module, we released findONTime (https://github.com/INSaFLU/findONTime) [28], which is a complementary tool designed to run concurrently to MinION sequencing towards a more timely and cost-effective real-time metagenomics virus detection using the INSaFLU-TELEVIR platform. Indeed, by automating the input preparation (ONT reads and metadata) and TELEVIR deployment, findONTime potentiates the detection of a virus in a sample as early as possible during the sequencing run, reducing the time gap between obtaining the sample and the diagnosis, and also reducing sequencing costs (as ONT runs can be stopped at any time and the flow cells can be cleaned and reused). As a proof-of-concept exercise, we ran the findONTime over ONT data of a MPXV-positive sample (regarding the first 2022 outbreak genome described in Isidro et al. (2022)) [8] that was subjected to MinION shotgun metagenomics after DNA extraction without any virus enrichment / host-depletion laboratory treatment. As shown in Fig. 5, simulating a context of hypothesis-free ONT sequencing, this approach would allow us to get early sequence evidence for a rapid, robust, and less costly diagnosis. Indeed, although the proportion of MPXV reads was no more than 1%, strong sequence evidence was reached in less than 2 h, namely MPXV classification in both reads and contigs just after 40 min or more than 90% of MPXV reference genome covered by at least one read at 1 h 20 min of run time. findONTime can be used as a “start-to-end” solution or for particular tasks (e.g., merging ONT output files, metadata preparation and upload to a local INSaFLU-TELEVIR instance). Usage examples are provided in https://github.com/INSaFLU/findONTime#usage [28].
Fig. 5
Rapid, robust, and cost-efficient diagnostics using findONTime in combination with MinION sequencing. A simulated scenario of hypothesis-free ONT sequencing using data from a MPXV-positive sample, prepared without prior viral enrichment/host depletion. The plot shows the number of reads mapping to a MPXV reference genome and the percentage of horizontal coverage at increasing time points, during the sequencing run. Reference genome identified with over 50% coverage after 20 min. Contigs mapped at the 40-min mark. Strong evidence (mapped contigs; > 90% reference genome covered by at least one read) is achieved in under 2 h (1 h 20 min)
Routine genomic surveillanceThe surveillance-oriented component of the platform dashboard is divided into:
1.INSaFLU Projects—from reads to reference-based generation of consensus sequences and mutation annotation/screening, followed by gene- and genome-based alignments, amino acid alignments, classification, NextClade link, etc.
The COVID-19 pandemic and other recent international public health threats (e.g., the multi-country mpox outbreak, the A/H5N1 avian influenza global spread, etc.) have contributed to accelerate the “universal” access to modern sequencing technologies, in particular to portable third-generation sequencing equipments (MinION). As such, to keep following this technological revolution in the field of genomic surveillance of viral diseases, we have put particular efforts to improve and adapt the surveillance-oriented component of the INSaFLU-TELEVIR platform so that it could handle ONT sequence data of multiple viruses (besides Illumina and Ion Torrent data, as described in the first release [19]). Similarly to the Illumina / Ion Torrent pipeline, the developed ONT pipeline incorporates software for reads quality control, reference-based mapping, primer clipping, mutation calling, and consensus generation, and performed similarly to the widely used ARTIC SARS-CoV-2 pipeline (https://github.com/artic-network/fieldbioinformatics/) [53], as detailed in the benchmarking results of the Implementation section. We privileged a very smooth integration of the new ONT pipeline into the existing dashboard [19] by keeping the same user interface and features as for the existing pipeline, in order to minimize the impact on its usability and promote data analysis flexibility (e.g., ONT and Illumina samples can be run in the same project). In brief, the updated INSaFLU projects can process samples from the different sequencing technologies, which are automatically detected upon reads upload and automatically guide the pipeline to be run, without further user interaction. All upstream INSaFLU analyses (e.g., mutation annotation, alignments, and phylogenetics) and outputs (content and format) (e.g., tabular list of mutations and its annotation) were kept similar to the existing Illumina/Ion Torrent pipeline in order to facilitate sequence comparison regardless of the technology used. This harmonization and flexibility is particularly useful, for instance, in the context of routine genomic epidemiology systems with centralized data analysis, but decentralized sequencing with distinct technologies.
In addition to the integration of the reference-based genome assembly pipeline for ONT data, the INSaFLU projects were upgraded with other important surveillance-oriented (often virus-specific) functionalities and features, including (i) integration of automatic SARS-CoV-2 Pango lineage assignment (https://cov-lineages.org/pangolin) [78] using Pangolin (https://github.com/cov-lineages/pangolin) [55, 56, 79]. To better fit this dynamic lineage nomenclature, whenever new software/database versions are released (automatically checked daily), a button “Update Pango lineage” is automatically made available, so that users can re-assign all project samples using the latest software/database versions; (ii) integration of direct links to Nextclade (https://clades.nextstrain.org/) [58] for rapid and flexible SARS-CoV-2, seasonal influenza, MPXV and RSV consensus sequences analysis (at client side on browser). This feature allows INSaFLU-derived consensus sequences to be easily subjected to quality screening, lineage/clade/genotype classification, mutation exploration and other relevant analyses available at the Nextclade framework; (iii) incorporation of the newly developed “algn2pheno” (see implementation) for automatic screening of SARS-CoV-2 Spike amino acid alignments against “genotype-phenotype” databases of mutations of potential biological or epidemiological interest; (iv) improvement of existing features for phylogenetic trees visualization using PhyloCanvas (https://github.com/phylocanvas) [80] to easily color tree nodes and to display colored metadata blocks next to the phylogenetic trees nodes, thus facilitating integration of relevant epidemiological and/or clinical data and pathogen genomic data; and (v) inclusion of novel “expand-and-collapse” panels for an interactive report of all detected mutations (including detailed information about genome position, nucleotide change, coverage evidence, frequency, and impact at protein level), the mean depth of coverage and horizontal coverage per locus for all samples through intuitive color-coded buttons and an “algn2pheno” report of mutations of interest.
2.Nextstrain Datasets—from consensus sequences to advanced Nextstrain phylogenetic and genomic analysis, coupled with geographic and temporal data visualization and exploration of sequence metadata.
The Nextstrain (https://nextstrain.org/) [59, 60] project has played an important role in harnessing the scientific and public health value of pathogen genome data in the prevention and control of infectious diseases (well demonstrated during the COVID-19 pandemic), but also by providing up-to-date analyses of virus evolution at a global scale as well as open-sourced analytic and visualization tools. In this context, in order to promote and facilitate the real-time tracking of virus evolution (from NGS reads to the tip of the tree), we strengthened the genomic surveillance component of the INSaFLU-TELEVIR platform by integrating Nextstrain workflows for advanced analysis, visualization, and exploration of phylogenetic and genomic data together with geographic and temporal data (or any other epidemiologically relevant metadata variable). We provide the functionality of Nextstrain workflows as a new type of project named “NextStrain Dataset”. Upon creation of a new dataset, the user selects a specific Nextstrain build, either a virus-specific build (available for the four seasonal influenza, avian influenza, SARS-CoV-2, MPXV, and RSV A/B, at the time of publication) or a “generic” build that can be used for other viruses (see Implementation). For instance, a TELEVIR partner (INIA) has successfully tested the generic build with West Nile Virus data, showing its applicability to several viral threats. After creation, users can then select samples to be included in the dataset from three sources. The most common origin of the samples is reference-based assembly projects (classical INSaFLU projects), from which generated consensus sequences and associated sample metadata are automatically sent to the dataset. Users can also import sequences from the References repository (especially useful when using the “generic” build) as well as externally-provided sequences (directly uploaded as single or multi-fasta files). In the latter cases, since there is no associated metadata, default values are assumed for build-specific mandatory metadata parameters (e.g., the collection date is defined as the current date). Still, at any time, users can download the automatically generated Nextstrain metadata table, and update the default values by uploading a modified metadata file (as a tabular tsv file). To take advantage of temporal and geographical features of Nextstrain and increase their robustness, users must provide (1) “date” for all samples added to Nextstrain datasets—if no collection date is provided, INSaFLU will automatically insert the date of the analysis as the “collection date”, which might (considerably) bias (or even break) the time-scale trees; (2) “latitude” and “longitude” and/or “region”, “country”, “division” and/or “location” columns in the metadata—these values are screened against a database of geographical coordinates to geographically place the sequences in the Nextstrain map. When all samples are imported, and metadata is up to date, the user can then (re-)run the analysis and download the input consensus sequences (as a fasta file) and metadata table, as well as outputs from the build process, such as nucleotide alignments (as a fasta file), the divergence tree (as a newick file) and json file(s) that can be client-side visualized using auspice (https://auspice.us/) [81]. Consensus sequences imported into Nextstrain datasets can also be directly sent to Nextclade.
ImpactSince its first release [19], the INSaFLU (https://insaflu.insa.pt/) [22] bioinformatics framework, which has been considerably upgraded as described in the present study, has played a pivotal role in pathogen genomics surveillance in Portugal, namely for SARS-CoV-2 (https://insaflu.insa.pt/covid19/; more than 48,000 sequences analyzed, as of October 2023) [
Comments (0)