
Remember me
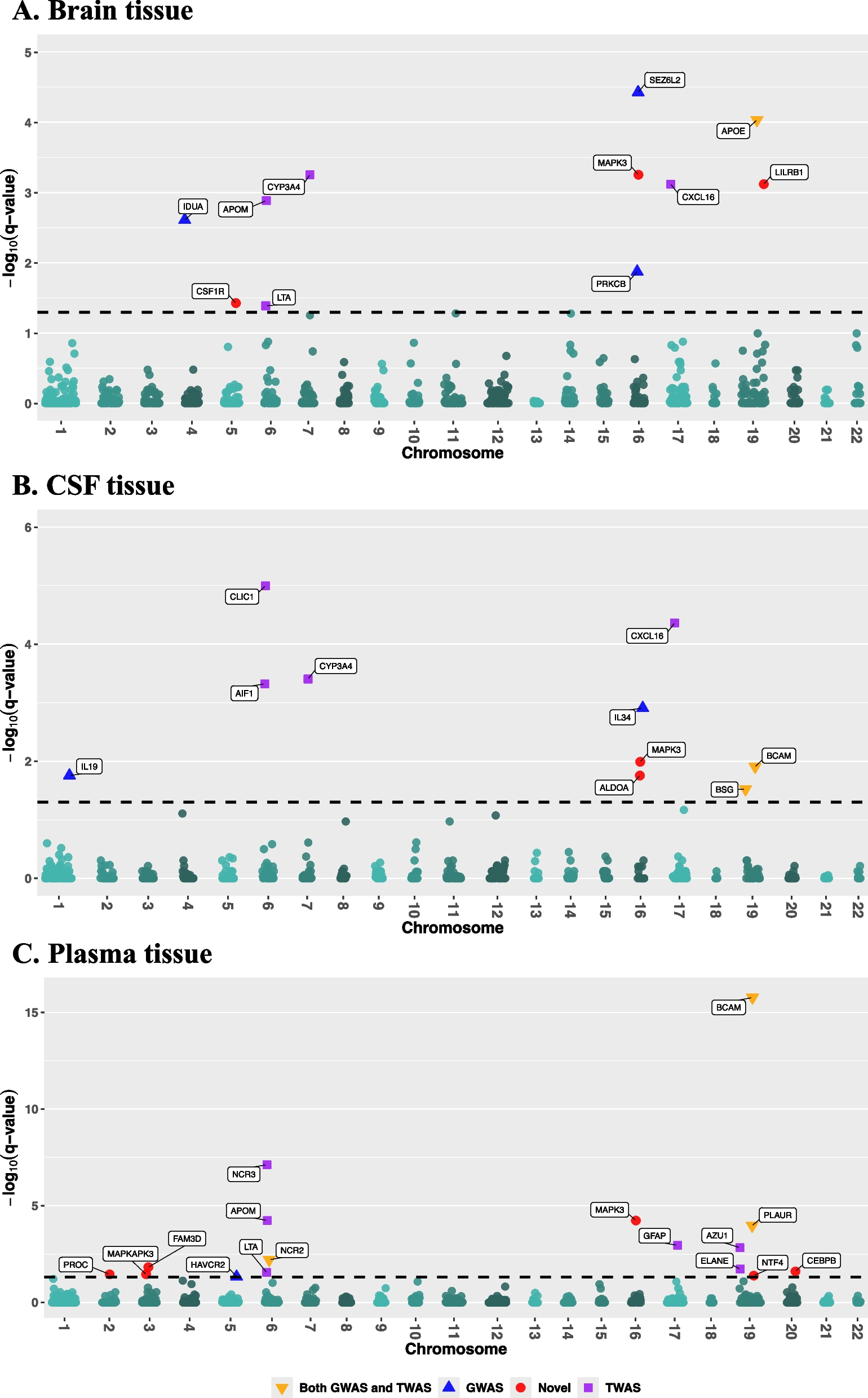





Data presented from the Core includes 1795 participants from Clarity AD double blind, 897 randomized to placebo and 898 randomized to lecanemab. Data from the Core + OLE includes 1612 participants with at least one dose of lecanemab, 898 participants randomized to lecanemab in the Core and 714 participants who received placebo in Core and then converted to lecanemab in OLE. Of the 1612 lecanemab-treated participants, 1321 had exposure of greater than or equal to 6 months, 1007 participants had exposure of greater than or equal to 12 months, 505 participants had exposure greater than or equal to 24 months and 47 participants had exposure of greater than or equal to 36 months in this data cut off (as of December 1, 2022).
Baseline characteristics for those in the Core + OLE were generally similar to the characteristics in both treatment groups of the Core study (Table 1). The Core + OLE population had a mean age of 71.5 years, were 52.4% female, and 76.2% Caucasian. Overall, 69.3% of participants were ApoE ε4 carriers (53.8% heterozygotes; 15.4% homozygotes). In the Core + OLE, baseline antithrombotic use was 36.5% and the most common antithrombotic medication was acetylsalicylic acid (27.5% overall and 75.2% of those individuals with concomitant antithrombotic medication use). Overall, the Clarity AD study was conducted in patients with broad range of comorbidities and concomitant medications, from a diverse racial and ethnic background, and from clinical trial practice settings similar those for the general population.
Table 1 Characteristics of participants at baseline in the intent-to-treat populationGeneral safety updateGeneral safety outcomes for Core + OLE can be found in Table 2, which also include those from the previously published Core study for comparison [7]. In the Core + OLE, deaths occurred in 1.0% and SAEs were experienced by 15% of participants. The occurrences of deaths or SAEs were similar regardless of ApoE ε4 genotype (Table S2). In the Core, there were 7 deaths on placebo (0.8%) and 6 on lecanemab (0.7%), and none were considered related to study drug. Two additional deaths (1 placebo and 1 lecanemab) occurred 30 days after last study treatment administration in the Core. In the OLE treatment period, there were 9 additional deaths as of data cut off for this manuscript, with 4 deemed possibly related to study treatment. Of the 24 deaths in placebo or lecanemab treatment groups across the Core + OLE, 3 were due to intracerebral hemorrhage: 1 placebo in the Core due to intracerebral hemorrhage (ICH), and 2 lecanemab in OLE with concurrent ICH (1 after tissue plasminogen activator [tPA] and 1 on anticoagulant therapy). Exposure-adjusted rates of death for lecanemab in the Core + OLE was 0.0069 per participant year, which was similar to the rate for placebo in Core (0.0065 per participant year; Table S3). Additional narrative detail of all deaths and a summary of SAEs occurring during Core + OLE can be found in Table S4 and Table S5, respectively. The most common SAEs in the Core + OLE were infusion-related reactions (1.2%) and ARIA-E (1.1%).
Table 2 Adverse events and ARIA in Clarity Core and Core + OLEIn Core + OLE, 86.2% of individuals in Core + OLE had at least one adverse event. (Table 2). Treatment-related adverse events and adverse events leading to drug discontinuation occurred in 44.7% and 7.7% of participants, respectively. The most common adverse events (> 10%) were infusion-related reactions (24.5%), ARIA-H (18.5% [microhemorrhages and superficial siderosis]), COVID-19 (14.7%), ARIA-E (13.6%), and headache (10.3%).
ARIA-E and ARIA-H in Clarity ADARIA-E and ARIA-H were protocol-specified as adverse events of special interest in the lecanemab Clarity AD study and the top-level ARIA-E data for the Core study have been summarized previously [7]. ARIA-E and ARIA-H data for the Core and Core + OLE can be found in Table 2 and representative examples of imaging are shown in Figure S1. In Core + OLE, ARIA-E occurred in 13.6% of participants, with 3.3% symptomatic ARIA-E. When present, reported symptoms for participants with ARIA-E included headache, confusion, dizziness, vision changes, nausea, aphasia, weakness, or seizure. Focal neurologic deficits may also occur. Symptoms associated with ARIA-E usually resolve over time.
In Clarity AD, seizures were an infrequent symptom of ARIA-E or ARIA-H. In the Core + OLE, there were a total of 10 participants (0.6%) with seizures occurring concurrently with ARIA-E or ARIA-H (including ICH). Incidence of seizures unassociated with ARIA-E or ARIA-H events was similar (9/1612 [0.6%]); the incidence of unassociated seizures was the same between treatment groups: placebo 3/897 (0.3%) and lecanemab 3/898 (0.3%) in the Core study. Exposure-adjusted rates of death with concurrent ARIA (ARIA-E or ARIA-H; irrespective of whether ARIA-E or ARIA-H was the cause of death) were also similar between lecanemab in the Core + OLE and placebo in the Core (0.0013 [3 cases] and 0.0008 [1 case], respectively).
ARIA-E was more common in ApoE ε4 carriers, with highest frequency in homozygotes (noncarriers: 6.5%; heterozygotes: 11.6%; homozygotes: 34.5%). ARIA-E with lecanemab in Core + OLE generally occurred within the first 3 months (71%) or 6 months (92%; Fig. 1), generally resolving within 4 months of detection (81%), regardless of ApoE ε4 carrier status. Specifically, 60 of 111 participants (54%) were resolved by MRI at 90 days and 90 of 111 participants (81%) resolved by 120 days. Newly treated Core placebo participants had similar ARIA-E rates in the OLE as those for Core lecanemab participants.
Fig. 1
Timing of ARIA-E events (A) overall and (B) by APOE4 genotype for lecanemab in Core + OLE and placebo in Core
A summary of radiographic and clinical severity of ARIA-E overall and by ApoE ε4 genotype can be found in Table 3. ARIA-E events were primarily mild-to-moderate radiographically (93/218; 88.5%) and asymptomatic (96.7%), regardless of ApoE ε4 genotype. Overall, 51% (49/96) participants with mild radiographic ARIA-E at onset did not worsen and continued dosing without drug interruption. Participants with mild radiographic ARIA-E who continued dosing resolved (3 months) in a similar time frame to those who discontinued dosing (4–5 months). Serious symptoms associated with ARIA-E were reported in 0.7% (6/898) of participants treated with lecanemab. Clinical symptoms associated with ARIA-E resolved in 79% (23/29) of participants during the period of observation.
Table 3 ARIA-E events: radiographic and clinical severity overall and by APOE4 genotypeThe majority (> 95%) of participants had at least one post-ARIA cognitive assessment which was included in the primary analysis. Multiple analyses which included incorporating data after ARIA events (primary mixed model for repeated measures [MMRM]), censoring data after ARIA events, and imputing the data after ARIA events with placebo mean change show that ARIA did not adversely impact cognition or function. Results were highly statistically significant across all these analyses demonstrating that occurrence of ARIA events did not impact the efficacy of lecanemab. The impact of ARIA or ARIA-E on clinical efficacy was further evaluated by including ARIA or ARIA-E, as a [participant level and also as time-varying], covariate in the MMRM model for CDR-SB, Alzheimer’s disease Assessment Scale-Cognitive Subscale (ADAS-Cog14), and Alzheimer’s Disease Cooperative Study–Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL). ARIA or ARIA-E was not a significant covariate, and the estimated progression in those with or without ARIA or ARIA-E were similar.
The frequency of ARIA-H in Core + OLE was 18.5%, with symptomatic ARIA-H occurring in 1.7% of participants treated with lecanemab (Table 2). ARIA-H that did not occur together with ARIA-E (i.e., isolated ARIA-H) was 9.1% overall and 0.4% symptomatic. In the Core, isolated ARIA-H occurred at similar rates in the lecanemab and placebo groups [7] and results for Core + OLE were consistent. Timing of isolated ARIA-H events was at a steady rate across the treatment course at the same rate as placebo, while ARIA-H concurrent with ARIA-E, termed concurrent ARIA-H, occurred early in treatment. (Fig. 2). For ICH, there was likewise no clear relationship with timing of treatment initiation (Table 4). ApoE ε4 carrier status contributed to the incidence but not the timing of ARIA. Newly treated Core placebo participants had similar ARIA-H rates in the OLE as those for Core lecanemab participants.
Fig. 2
Timing of ARIA-H events (A) overall ARIA-H, (B) isolated ARIA-H, and (C) concurrent ARIA-H for lecanemab in Core + OLE and placebo in Core
Table 4 Intracerebral hemorrhage in lecanemab studiesFor severity, ARIA-H events were largely mild-to-moderate radiographically (240/298; 80.5%) and asymptomatic (271/298; 90.9%), with results consistent across ApoE ε4 genotypes. Most participants with mild radiographic ARIA-H did not worsen and could continue dosing without drug interruption (156/188 [83%]). Most cases of first mild radiographic ARIA-H (125/156, 80%) were stable at the next MRI. Severe ARIA-H in the Core + OLE was reported in 57 (3.5%) participants, mostly driven by any microhemorrhage event that resulted in a cumulative number greater than 10 microhemorrhages (47/1612 [2.9%]).
In Core + OLE, there was a low rate of ICH with lecanemab therapy (8/1612; 0.5%), which was higher than the rate observed in the Core placebo group (1/897; 0.1%). The rate of ICH for lecanemab-treated participants on anticoagulants was 2.7% (4/147) (Table 2). The background rate of ICH in AD participants on anticoagulation is not known but might be expected to be higher than in non-AD participants due to CAA; therefore, comparative risk is difficult to assess. There was no clear relationship of ICH to ApoE e4 status, baseline MRI, or timing of treatment.
Antiplatelet and anticoagulant utilization during Clarity AD is summarized in Table 5. In the Core, ARIA rates were slightly higher in the placebo group with anticoagulants, whereas ARIA rates were generally lower in participants who were on antiplatelet agents as well as those who were on anticoagulants. ARIA appeared less frequently in lecanemab-treated participants who were on antithrombotic medications, both antiplatelets and anticoagulants (participants who were on antithrombotic medications received similar overall lecanemab dose as those that were not). Results were generally consistent across ApoE ε4 genotypes and results in Core + OLE population were similar to Core results for lecanemab-treated participants. Logistic regression analyses were performed to evaluate the impact of various risk factors on ARIA-E, including baseline co-morbidities like hypertension, baseline amyloid status, APOE4 genotype, microhemorrhage at baseline, baseline disease stage etc. Based on these analyses, the only baseline risk factors identified for ARIA-E were ApoE ε4 genotype, microhemorrhage at baseline, and white matter abnormalities.
Table 5 Antiplatelet and anticoagulant use in Clarity AD in Core and Core + OLEInfusion reactionsInfusion-related reactions were limited in impact and most often occurred only at one infusion, without recurrence, regardless of whether prophylactic medications were administered. In the Core + OLE, infusion-related reactions occurred in 398/1612 (24.7%) of participants and in 142/714 [19.9%] of newly treated lecanemab participants in OLE. Infusion-related reactions were largely mild-to-moderate (96.5%) and largely occurred on the first dose (73%). Most participants (65.1%) with such reactions only had 1 infusion-related reaction, with common symptoms of infusion-related reactions including fever and flu-like symptoms (chills, generalized aches, feeling shaky, and joint pain), nausea, vomiting, hypotension, hypertension, and oxygen desaturation. Overall, 46.9% of participants received preventative (“prophylactic”) medications (e.g., acetaminophen, antihistamine, hydrocortisone) for an infusion after experiencing the first infusion-related reaction. Of the 173 participants who received at least 1 preventative medication, 68 (39.3%) had subsequent infusion reactions; 60.7% did not have subsequent infusion reactions. Of the 196 participants who did not receive a preventative medication, 64 (32.7%) had subsequent infusion reactions and 67.3% did not have subsequent infusion reactions. Severe infusion-related reactions (grade 3–4) occurred in 10/1612 (0.6%) participants, all of whom experienced the reaction with the first dose.
Comments (0)