Cell lines and culture conditions
The Chinese hamster ovary (CHO) cell line (ATCC, USA) was cultured in a 44% mixture consisting of Dulbecco’s modified Eagle’s medium (DMEM; Corning, 10-013-CVR) and Ham’s F-12K (GIBCO, 21127022). The culture media were further enriched with supplements including 10% fetal bovine serum (FBS; GIBCO, 10091148), 1% 200 mM L-Glutamine (GIBCO, 25030081), and 1% Hypoxanthine-Thymidine Supplement (HT Supplement; GIBCO, 11067030). For the cultivation of Jurkat-PD-1 (Shcell, China) cell line Roswell Park Memorial Institute (RPMI 1640; Corning,10-040-CM) supplemented with 10% FBS. Similarly, the 293T-CTLA-4 (Shcell, China) cell line was cultured in DMEM supplemented with the same amount of FBS. All tumor cells were detected by the PCR short tandem repeat (STR) method and screened for the presence of mycoplasma monthly. All these aforementioned cell lines were maintained with an incubator set to 37 °C under humidified conditions containing 5% CO2.
Cells electroporation and transfection
In accordance with the guidelines provided by the Human T Cell Nucleofector™ Kit (LONZA, VVPA-1002), we obtained a total of 1E5 CHO cells during the logarithmic growth phase and resuspended in Human T Cell Nucleofector buffer with 20 μg of mRNAs. To ensure optimal transfer efficiency, we used the appropriate power transfer program CHO-K1 (H-014) Following established protocols, supernatant samples were subsequently collected at specific points (24, 48, and 72 h post electroporation) for subsequent analysis.
For in vitro transfection, 1.2 mL of medium was added to each well of a 12-well plate (NEST, 712001) followed by the addition of the prepared LNP-mRNAs. After a transfection period of 24 h, the cell culture supernatants were collected for subsequent ELISA analysis.
LNP formulation and characterization
The LNPs were prepared following the procedure outlined in PCT/CN2022/118198. In summary, a lipid solution comprising an ionizable lipid, cholesterol, a phospholipid (DOPE), and DSPE-PEG200 (Shanghai Advanced Vehicle Technology L.T.D. Co) was prepared in ethanol. The molar composition employed was 35:46.5:16:2.5. To obtain an aqueous solution of mRNA, we diluted the mRNA in a 20 mM citrate buffer (pH = 6.1). Subsequently, liposomes were generated using a microfluidic device, by combining the ethanol lipid solution and mRNA solution at a volume ratio of 1:3 organic to aqueous phase, with a total weight ratio of approximately 10:1 for lipids to mRNA.
The LNP characterization was conducted using the Malvern Zetasizer Nano ZS (Malvern UK) with backscattering detection mode at a temperature of 173 °C. This method allowed for the measurement of the z-average diameter and polydispersity index (PDI) of the lipid nanoparticles through dynamic light scattering.
Surface plasmon resonance (SPR) analysis
SPR analysis was employed to determine the binding avidity of the nanobodies with PD-1 and CTLA-4. According to the human antibody capture kit manual (cytiva, BR100839), the IgG (50 μg/mL) were immobilized onto a CM5 sensor. Subsequently, purified nanobodies were applied onto the pre-coated chip surface. Gradient concentrations (200, 100, 50, 25, 12.5, 6.25 nM) of the nanobodies were introduced to the chip surface at a flow rate of 30 μL/min to facilitate antigen capture by the IgG. Regeneration of the chip was carried out by employing a flow rate of 30 μL/min and a second 10 mM Glycine-HCl at pH 2.0 for a duration of 30 s. The data were collected by Fortebio Data Acquisition 7.0. Subsequently, Biacore Evaluation Software 2.0 was utilized to analyze the bond rate (kon), dissociation rate (koff), equilibrium constant (KD), and to fit the obtained curves using the Langmuir 1:1 model.
High-throughput protein liquid chip
A high-throughput protein liquid chip, following the experimental techniques outlined by the PPX-06-MXZTFZG, was used to detect cytokines within the mouse tumor. The Luminex 200TM System was used to quantify the mean fluorescence value (MFI), while a five-parameter nonlinear regression was applied to fit the standard curve. The obtained MFI values were then converted to their corresponding concentrations based on the established standard curve.
Flow cytometry assay
Jurkat-PD-1 and 293T-CTLA-4 cell lines were incubated with bispecific nanobodies for 20 min at room temperature, followed by three washes with PBS. The washed cells were then stained with a MonoRab™ Rabbit Anti-Camelid VHH Cocktail conjugated with a phycoerythrin (PE) antibody (A02227, GenScript, China). The binding affinity of the bispecific nanobodies towards PD-1 and CTLA-4 on the cell surface was assessed using flow cytometry. Additionally, tumor microenvironment (TME) cells were surface-stained using fluorescently labeled antibodies targeting CD45(560510, BD Biosciences, USA), CD3e (557596, BD Biosciences, USA), CD8a (100762, Biolegend, California, USA), and CD335 (137611, Biolegend, California, USA). All samples were collected on Cytek (NC-CLC100, California, USA) or Beckman Coulter cytoFLEX (ONC-FCY-001, California, USA) and analyzed using Flow Jo software vX.0.7.
Enzyme-linked immunosorbent assay (ELISA)
The expression of bispecific nanobodies in culture supernatants and sera from mice was performed using the ELISA. A 96-well microplate was coated with Human PD-1 (Acro, H5221) at 4 °C overnight. A concentration response curve was established by diluting Z15-0 and Z15-0-2 standard from 12.5 ng/mL down to a 2× dilution, resulting in 7 gradients and a concentration of 0 ng/mL. After cleaning and closing the plate, a MonoRabTM Rabbit Anti-Camelid VHH Antibody (GenScript, A01861) was used as the detection antibody. The optical density (OD) values were measured at 450 nm using an EnVision system (PerkinElmer, Massachusetts, USA), and the curve of OD and antibody concentration was fitted using a Four Parameter Logistic (4PL) model.
Mixed lymphocyte reaction (MLR)
Human peripheral blood mononuclear cells (PBMCs) were obtained from two healthy donors sourced from Shanghai Aoneng Biotechology Co., Ltd. (Shanghai, China) through Ficoll-Paque density gradient centrifugation. PBMCs from one donor were used for the preparation of CD4 + T cells, while the PBMCs from the other donor were used for DC induction. The specific procedure for DC induction can be found in CN107574149B. Additionally, CD4+ T cells were isolated from a separate donor’s PBMCs following the instructions provided by the manufacturer of the CD4+ T Cell Isolation Kit (Milteniy Biotech, Bergisch Gladbach, Germany).
During the experiment, mature dendritic cells (DCs) were washed with DPBS (Hyclone, SH30028.02) and then mixed with 2E5 mDCs in Opti-MEM™ (GIBCO, 31985070) medium within 24-well plates (NEST, 702001). The LNP-encapsulated mRNA was added at a concentration ranging from 0 to 2000 ng per well. The plate was then incubated at 37 °C with 5% CO2 for 4 h. Following this incubation period, 8E5 freshly isolated CD4+ T cells were added to each well and co-cultured for 5 days. After incubation, the culture supernatants were collected and assessed for the secretion of IL-2 using the Human IL-2 ELISA Kit (Sino Biological, KIT11848) following the provided manual instructions.
In vivo experiments
All mice aged 6–8 weeks were obtained from PharmaLegacy (Shanghai, China) and housed in IVC Sealsafe cages under a 12/12 light/dark cycle at a temperature range of 20-26 °C. The animal-related experiments conducted in this study received approval from the Experimental Animal Ethics Committee of Shanghai University. Investigators were not blinded during data collection or analysis. Experimental and control animals were treated equally.
The C57BL/6 mice were used for pharmacokinetic evaluation, where LNP-bispecific nanobodies mRNA was intravenously (i.v.) administered at doses of 0.2 and 1 mg/kg through their tail veins. Blood samples were collected from the orbit and the serum was extracted for ELISA analysis.
The hCTLA-4/hPD-1 transgenic C57BL/6 mice were received subcutaneous injections of 1E6 MC38 tumor cells. Tumor volume (TV) was measured biweekly using calipers and the formula (width2 × length)/2. Once successful tumor engraftment occurred, with an average tumor volume of 72.78 mm3, the mice were randomly divided into groups receiving either administered subcutaneous LNP-mRNAs or bispecific nanobodies. To evaluate tumor growth inhibition (TGI), we employed a calculation method represented by (1 − T/C) × 100%, where T represents relative tumor volume in the experimental group compared to the control group at a specific time point denoted by C; meanwhile, T/C % indicates relative rate of tumor growth progression over time period examined. At the end of the experiment, all tumors were collected and weighed for further analysis purposes.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism software version 9.5.1. One-way analysis of variance (ANOVA), Student’s t-test and Tukey’s multiple comparison test was used to determine the statistical significance, with a P-value threshold of less than 0.05. The sample size for in vivo experiments was based on previous similar experiments in our laboratory. For all other studies, the data were replicated more than three times to ensure the robustness and reliability of the findings in line with rigorous scientific standards.

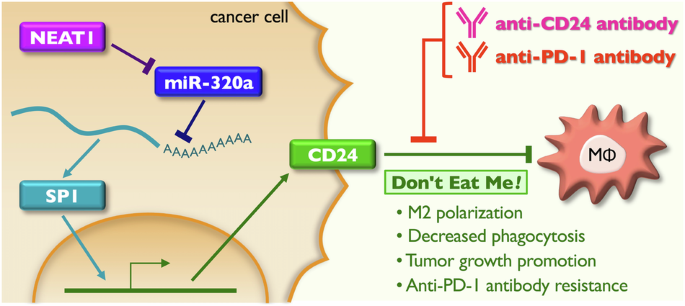
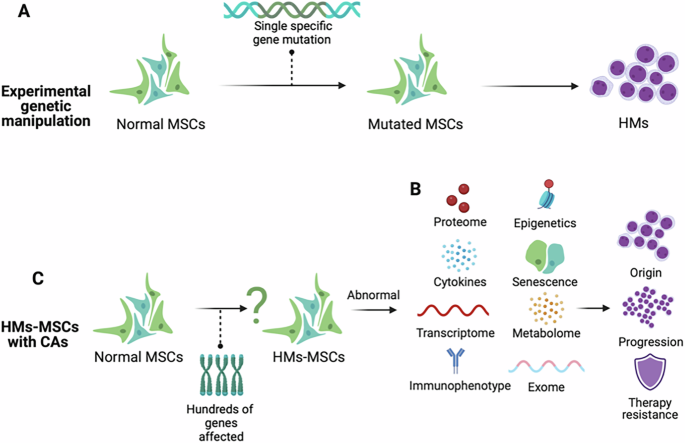
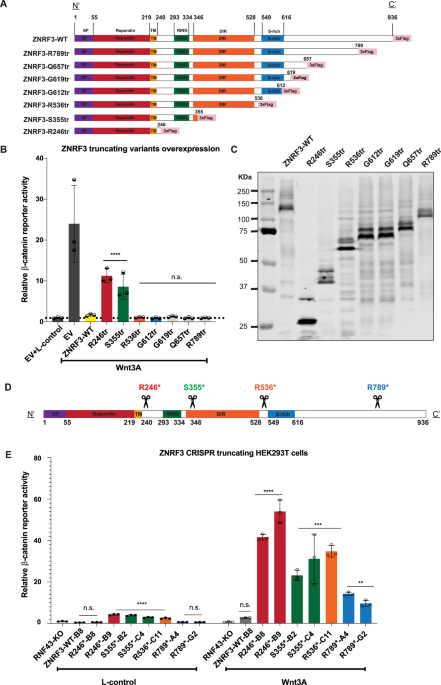
Comments (0)