
Remember me



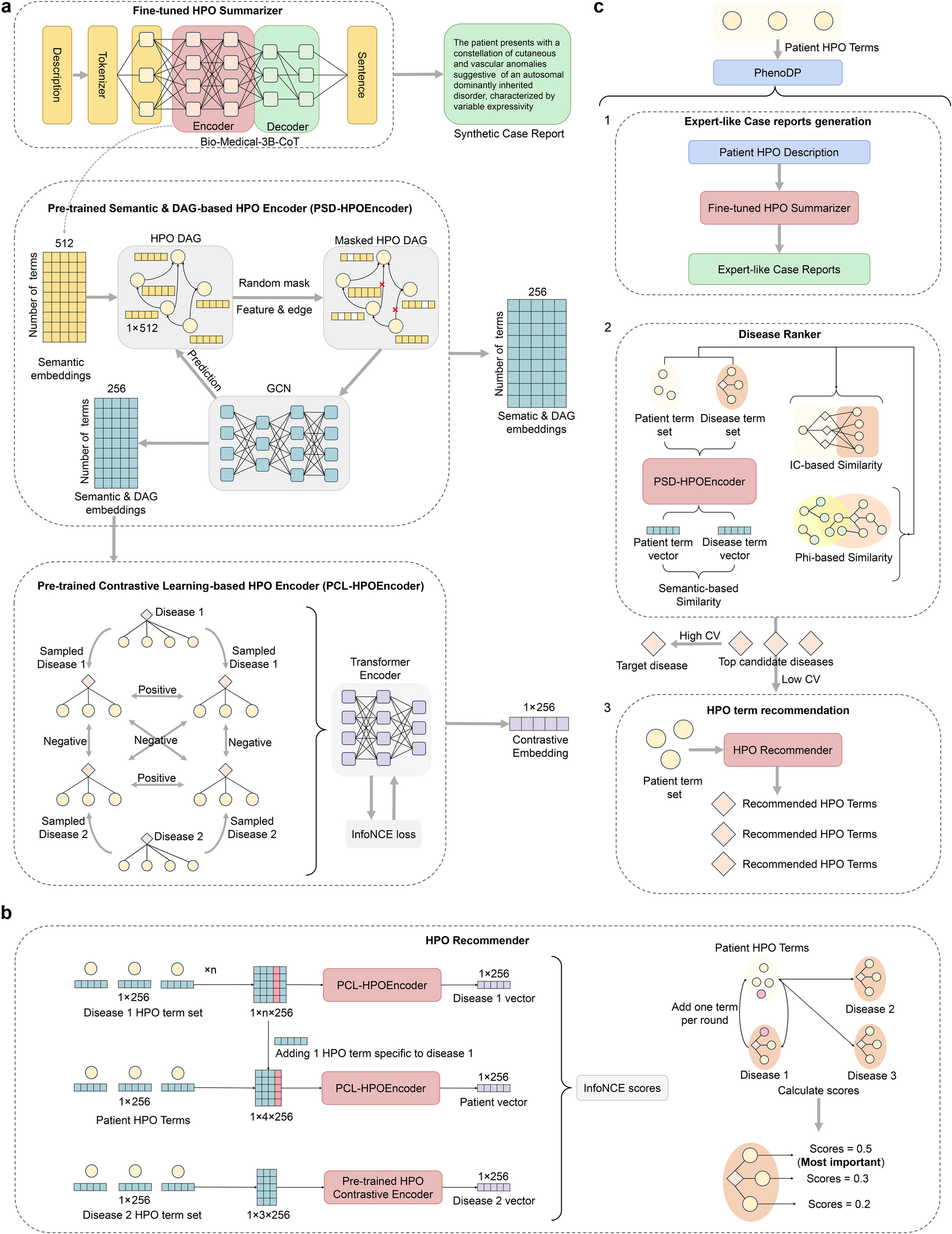



The design of this study is illustrated in Fig. 1. The objective of the present study was to determine and evaluate the predictive performance of an integrated risk score (IRS), which encompasses genetic and conventional risk factors, for CVD and related cardiometabolic outcomes in African populations. The genetic factors in the IRS consist of multiple PGS and genetic ancestry, as inferred by projected principal components of ancestry (PPCs). Conventional risk factors, referred to as non-genetic factors going forward, included sociodemographic and lifestyle risk factors such as age, sex, smoking status, alcohol consumption, diet-related factors, physical activity, and nightly sleep duration.
Fig. 1
Study design and overview. This study employed two primary datasets, the base and target datasets. Base: The African Partnership for Chronic Disease Research (APCDR) dataset, encompassing 14,126 participants from South Africa, Kenya, Uganda, Ghana, and Nigeria, and the target was the Africa Wits- INDEPTH Partnership for Genomic Research (AWI-Gen) dataset which included 10,602 participants from Burkina Faso, Ghana, Kenya, and South Africa. Polygenic scores for 14 cardiometabolic traits were derived using the p-value thresholding method combined with linkage disequilibrium (LD) clumping. The African subset of the 1000 Genomes Project (1 KG) served as the reference panel. Predictive modelling evaluated the efficacy of genetic, non-genetic, and integrated models in forecasting disease outcomes. The map included in the figure visually represents the approximate geographical distribution of the cohorts, with the position circles indicating the location. Key acronyms: APCDR, African Partnership for Chronic Disease Research; AWI-Gen, Africa Wits- INDEPTH Partnership for Genomic Research; CVD, cardiovascular disease; GWAS, Genome-Wide Association Study; LD, Linkage disequilibrium; PGS, Polygenic score
Summary statistics from the African Partnership for Chronic Disease Research (APCDR) GWAS meta-analysis for cardiometabolic traits were used to derive PGS. The APCDR cohort consists of 14,126 individuals from different African regions [18]. Scores were trained on data from the Africa Wits-INDEPTH Partnership for Genomic Research (AWI-Gen) [19] cohort comprising 10,603 individuals from four African countries. Study participants from AWI-Gen resided in Burkina Faso, Ghana, Kenya, and South Africa, while those from ACPDR were from Ghana, Kenya, Nigeria, South Africa, and Uganda. Despite a similar regional mix, there was no recorded sample overlap between the target cohort and the individuals analysed in the GWAS from which summary statistics were obtained.
First, we constructed distinct PGS for each of the fourteen cardiometabolic traits, all continuous phenotypes, using the p-value thresholding and LD clumping approach (pT + clump), and according to standard procedures outlined by Choi and colleagues [20], employing the GenoPred pipeline (https://github.com/opain/GenoPred/tree/master/GenoPredPipe) [21]. Scores were derived for the following traits: six anthropometric indices (body mass index (BMI), height, weight, hip circumference, waist circumference, and waist-to-hip ratio (W–H ratio)); two blood pressure measurements (diastolic blood pressure (DBP) and systolic blood pressure (SBP)); four lipid traits (low-density lipoprotein cholesterol (LDL), high-density lipoprotein (HDL), total cholesterol (TC) and triglycerides (TG)); and two liver function measures (albumin and bilirubin blood serum levels).
Subsequently, the associations of the PGS and non-genetic factors with thirteen of the cardiometabolic traits (excluding bilirubin, which was not measured in AWI-Gen) and seven CVD-associated outcomes, namely CVD, diabetes mellitus, dyslipidaemia, heart attack, hypertension, obesity, and stroke, were tested. Next, to derive and evaluate the predictive utility of genetic, non-genetic, and IRS models, elastic net regression with nested cross-validation (NCV) was used.
Data sourcesBase dataset: APCDR meta-analysisThe APCDR (African Partnership for Chronic Disease Research) is a genome-wide association meta-analysis of association statistics that encompasses association statistics derived from four African cohorts [18]. The cohorts included the Uganda Genome Resource (UGR) (n = 6188), the Africa-America Diabetes Mellitus Study (AADM) (n = 5231), the Durban Diabetes Study (DDS) (n = 1165), and the Durban Case Control (DCC) (n = 1542) [18, 22, 23]. In brief, the meta-analysis, as conducted by Gurdasani et al. (2019), investigated 34 cardiometabolic traits in up to 14,126 individuals aged 18 years and older residing in Ghana, Kenya, Nigeria, South Africa, and Uganda [18]. The APCDR data is publicly available and includes imputed dosage data for all individuals and ~ 96 million variants. These data were generated using METASOFT [24] with a composite reference panel developed by authors [18]. Summary statistics were downloaded from the NHGRI-EBI GWAS Catalog [25] on 01 Jun 2021 for studies GCST009042 to GCST009060 (details of studies are provided in Table S1) [18].
Target dataset: AWI-GenAWI-Gen is a cross-sectional cohort study undertaken across four sub-Saharan African countries: Burkina Faso, Ghana, Kenya, and South Africa [19]. This study’s primary objective is to explore genetic and environmental factors associated with cardiometabolic diseases in Africans. It is part of the Human Heredity and Health in Africa Consortium (H3Africa). From 2012 to 2016, approximately 12,000 participants, primarily between the ages of 40 and 60 years, were enrolled across six study centres, and individual-level genetic, health-related, and phenotypic data relating to lifestyle was collected. Baseline data was used in this study. The study sites are from South Africa, the MRC/Wits Agincourt Health and Demographic Surveillance System Site (HDSS) (referred to as Agincourt), the Dikgale HDSS of the University of Limpopo, and the Soweto Centre which is coordinated by the South African Medical Research Council/Wits Developmental Pathways for Health Research Unit (DPHRU); in Kenya, the African Population and Health Research Center HDSS in Nairobi; in Ghana, the Navrongo HDSS in the Navrongo Health Research Centre; and in Burkina Faso, the Nanoro HDSS hosted by the Institut de Recherche en Sciences de la Santé Clinical Research Unit [19, 26].
Genetic dataApproximately 11,000 individuals were genotyped on the 2.3 M SNP H3Africa array at Illumina® FastTrackTM Microarray services (Illumina, San Diego, USA). Genotype calling was performed using the Illumina pipeline. Quality control (QC) was performed as described previously, but in summary, pre-imputation QC was performed using the H3ABioNet/H3Agwas pipeline (https://github.com/h3abionet/h3agwas) and variants with a minor allele frequency (MAF) < 0.01, missingness > 0.05 or Hardy–Weinberg equilibrium p-value < 1 × 10−3 were removed. Additionally, SNPs from the X and Y chromosomes, mitochondrial SNPs, and SNPs that did not match the GRCh37 reference alleles were removed. Samples that were potential duplicates (PIHAT > 0.9), had a missing SNP genotyping rate greater than 0.05, and reported vs. genetic sex inconsistencies were excluded. Population stratification was assessed using principal component (PC) analysis based on an LD-pruned subset of SNPs using the smartPCA program implemented in EIGENSTRAT. Imputation was performed using the African Genome Resources reference panel (EAGLE2 + PBWT pipeline) at the Sanger Imputation Server (https://imputation.sanger.ac.uk/). Post-imputation QC involved removing indels, rare SNPs (MAF ≤ 0.01), and poorly imputed SNPs (Info score ≤ 0.6), resulting in a final dataset containing 10,603 participants and 13.98 M SNPs.
Phenotypic dataIn addition to demographic, general health and infection history variables, the AWI-Gen questionnaire provided information on diet, smoking status, alcohol use, physical activity, and sleep. The variables associated with CVDs and used in conventional CVD risk calculators were included in our models and referred to as non-genetic factors throughout our analyses [3, 27, 28]. The variables selected included age, sex, current smoking status, alcohol consumption status, sleep (hours/night), moderate and vigorous physical activity (minutes/week), juice (number per week) and sugar drinks (number per week). Current smoking status was obtained from “Yes”, “No” responses to the following question, “Do you currently smoke tobacco?” Similarly, alcohol consumption status was determined from “Yes,” “No” responses to the following question “Are you a current alcohol consumer?” Those who preferred not to answer or did not know, were excluded. The Global Physical Activity Questionnaire (GPAQ) was used to obtain self-reported physical activity. Total moderate-vigorous physical activity (MVPA) in minutes per week was calculated from the accumulation of occupation, travel-related and leisure time physical activity. Sitting time (minutes/week) is used as a proxy for sedentary behaviour [29]. Weekly consumption of bread (slices per week), fruit (servings per week), and vegetables (servings per week) was calculated by multiplying the individual’s number of servings per day by the number of times a week each respective food group was consumed. Not all the selected variables were available in the Soweto sample; thus, these samples were excluded from the prediction modelling analyses. For the remaining participants, individuals with more than 5% of the data missing among these selected variables were removed. Additional details on the variables and their construction can be found in Supplementary Materials S2 and S3.
Disease outcomesThe outcome variables in this study included 13 cardiometabolic traits and seven CVD-associated outcomes. Cardiometabolic traits included BMI, height, weight, hip circumference, waist circumference, and W–H ratio, DBP, SBP, LDL, HDL, TC, TG, and albumin and bilirubin blood serum levels. CVD-associated disease outcomes assessed were CVD, diabetes mellitus (T2D), dyslipidaemia (DLD), heart attack (HA), hypertension (HTN), obesity (OBS), and stroke. CVD was defined as present if the participant reported having had a heart attack, stroke, or transient ischaemic attack. Participants previously diagnosed with congestive heart failure or angina were also classified as having CVD. Transient ischaemic attack, congestive heart failure and angina outcomes are not included as single disease endpoints in our analyses due to the small sample size. Further information regarding the outcome definitions can be found in Supplementary Materials S2 and S3. For disease traits, all cases were included. The maximum sample sizes for each phenotype in the prediction modelling analyses are shown in Table 1.
Table 1 Sample sizes for each disease outcome (e.g., characteristics of the AWI-Gen cohort for the CVD traits and associated outcomes)NA refers to participants who reported that they did not know their disease status, and those from the Soweto site—these participants were excluded from modelling analysis given the high level of missingness of non-genetic risk factors. CVD includes heart attack, stroke, or transient ischaemic attack, and participants previously diagnosed with congestive heart failure or angina. Limited case numbers for transient ischaemic attack, congestive heart failure and angina restricted their use as disease endpoints themselves. Some individuals experienced multiple CVD outcomes, and subsequently, a summation of individual outcomes does not equate to the total number of CVD cases reported.
Polygenic scoringQuality control of datasetsQC of base data: GWAS summary statistics.
GWAS summary statistics of traits for inclusion in the study were selected due to their relevance to cardiometabolic disease, presence in the AWI-Gen cohort, as well as sharing a similar ancestry to the target AWI-Gen population. The identified summary statistics underwent a series of standard quality control (QC) procedures [20], including the extraction of HapMap3 variants, and the removal of ambiguous variants, or where variants had missing data. Variants were flipped to match the 1000 Genomes Phase 3 (1 KG) reference, and then variants were retained if the MAF > 0.01 in the African subset of 1 KG (1 KG AFR), the MAF > 0.01 in the GWAS sample, and the INFO > 0.6. GWAS summary statistics variants and samples were removed if they (1) had a discordant MAF (> 0.2) between the reference and GWAS sample, (2) had reported p-values outside the range of 0 to 1, (3) were duplicates, or (4) had a sample size > 3 SD from the median sample size.
QC of target data: ancestry classification.
Individuals in AWI-Gen were assigned to the five super populations present in the 1000 Genomes phase 3 (1 KG) reference sample [30], namely European, East Asian, South Asian, African, and Admixed American. Super population membership was predicted using a 1 KG reference trained elastic net model consisting of the first six reference-projected genetic principal components (PPCs). Principal components were defined in the 1 KG reference using HapMap3 SNPs in common between the 1 KG and AWIGEN data with a minor allele frequency > 0.05, missingness < 0.02 and Hardy–Weinberg p-value > 1 × 10−6. LD pruning for independent variants was performed in PLINK [31] after the removal of long-range LD regions [32], using a window size of 1000, step size of 5, and r2 threshold of 0.2.
A multinomial elastic net model, created using the “glmnet” R package [33], predicted super population membership in the 1 KG reference with fivefold cross-validation. This model, along with reference-derived principal components, was applied to AWI-Gen for similar predictions. Participants with a predicted probability over 0.5 were assigned to a super population, with all being assigned to the AFR superpopulation as expected.
Score constructionTypically, a PGS follows the form β1X1 + β2X2 + … + βkXk + … + βnXn, where βk represents the effect size attributed to each allele for a given cardiometabolic trait associated with SNP k. Xk is the number of effect alleles at SNP k, and n is the total number of SNPs in the PGS. To derive the PGS for each trait, we used (1) publicly available GWAS summary statistics described in Gurdasani et al. (2019); extracting the disease-associated variants, the effect allele, the estimated β-coefficient for the effect allele, and the p-value of each genetic variant, and (2) linkage disequilibrium (LD) between genetic variants from the African 1 KG LD reference panel (661 Africans) [30]. Scores were derived using the pT + clump approach. The pT + clump method is a robust approach that enhances the accuracy and relevance of PGS by selecting the most informative genetic variants while reducing redundancy due to LD. We used default LD-based clumping parameters (r2 = 0.1, window = 250 kb) to retain only the single most significant variant within each locus, as overly aggressive LD thresholds can detrimentally affect the predictive power PGS [20]. The 1 KG AFR was used to estimate LD. Ten p-value thresholds were considered (1 × 10−8, 1 × 10−6, 1 × 10−4, 1 × 10−2, 0.1, 0.2, 0.3, 0.4, 0.5 and 1). Polygenic scores were then calculated in AWI-Gen participants, imputing missing variants using the 1 KG AFR allele frequency. In the AWI-Gen sample, 1,104,4026 HapMap3 variants were present. Polygenic scores were standardised (scaled and centred) based on the mean and SD of PGS in the 1 KG AFR reference sample. The score calculations were performed using PLINK v1.9 as implemented in the GenoPred pipeline (https://github.com/opain/GenoPred).
Association testingFollowing PGS development, regression analysis was used to assess the within- and cross-trait predictive utility of each PGS, and with the seven disease outcomes of interest, while accounting for confounders such as age, sex, and the first eight with-in sample principal components to avoid PGS associations being confounded by population structure [15]. Similarly, regression analyses were run to assess the association between conventional risk factors, including age, smoking status, alcohol consumption, diet-related variables, sleep, and physical activity, with selected disease outcomes. Given the differences by sex of these traits within African populations, the analyses were adjusted for sex [34,35,36]. The proportion of variance for a trait explained by the PGS and non-genetic factors was computed as the phenotypic variance explained, R2. For PGS association testing, R2 was obtained from a full model including both PGS and covariates (PCs, sex, age, and age-squared) minus the R2 obtained from a model including covariates alone. R2 was not adjusted to the liability threshold model due to limited disease prevalence estimates available across Africa and the substantial variation in prevalence noted across cohort sites. For multiple testing, results were corrected for the number of PGS tested for each outcome (i.e., applying a p-value threshold of 0.05/14). We did not correct for the number of p-value thresholds as they are correlated, and a Bonferroni correction would be overly conservative. The performance of each PGS was assessed as the Pearson correlation (r) between the observed and predicted outcome values and the Area under the receiver operating characteristic curve (AUC) statistics calculated. Correlation was used as the main test statistic as it is applicable for both binary and continuous outcomes and standard errors are easily computed.
Derivation of genetic ancestry predictorsIn addition to PGS, reference-projected genetic principal components (PPCs) were included in prediction models to enhance prediction. Genetic principal components capture major axes of genetic variation, which primarily represent differences in genetic ancestry [37] and can be used to enhance prediction over PGS alone [38]. To prevent overfitting, the principal component SNP-weights should be derived independently of the target sample. Therefore, we used the PPCs described in Sect. 2.3.1.2, where the first six genetic PCs were derived from the 1 KG reference, and then projected these PCs into the AWI-Gen target sample.
Integrated risk score and prediction modellingElastic net regression with nested cross-validation (NCV) (https://github.com/opain/GenoPred/blob/master/Scripts/Model_builder/Model_builder_V2_nested.R) was used to develop and evaluate the predictive utility of three risk prediction models: genetic, non-genetic, and integrated:
a.Genetic:
i.MultiPGS—assessed the predictive utility of utilising multiple PGS compared to single-trait PGS
ii.MultiPGS + Ancestry—assessed the predictive utility of utilising multiple PGS and information relating to ancestry, specifically projected principal components.
b.Non-genetic—assessed the predictive utility of selected conventional risk factors.
c.Integrated—assessed the predictive utility when combining all genetic (MultiPGS and Ancestry) and non-genetic predictors.
Elastic net balances feature selection and regularisation to reduce over-fitting and address collinearity among predictors. It combines the properties of both ridge and lasso regression, where similar to ridge regression, elastic net applies a penalty to model coefficients which shrinks them towards zero, thus reducing the impact of less important predictors. And similar to lasso regression, elastic net performs variable selection by setting some coefficients to zero. By balancing the weight of ridge and lasso penalties, this regularisation removes the need for manual selection of predictors and selects and weights the most predictive variables appropriately, reducing redundancy and enhancing model interpretability [39]. NCV repeatedly partitioned the dataset into training, validation, and testing sets, and consisted of 5 outer folds with a 90–10 data split (90% training, 10% testing) to provide an unbiased estimate of the predictive utility of the model, and 10 inner folds (80% training, 20% testing) for hyperparameter tuning. The proportion of variance explained by a model was computed as R2. Hyperparameters were determined using the “caret” R package, which optimises the RMSE for continuous outcomes and accuracy for binary outcomes.
The predictive utility of the models were defined as the correlation between observed and predicted values of each model, and the comparative performance of the models assessed using William’s test (also known as the Hotelling–Williams test) as implemented by the “psych” R package’s “paired. r” function. The code used to prepare data and conduct analyses is available on the GenoPred Pipeline GitHub page (see Data and Code Availability).
For genetic (the MultiPGS and MultPGS + PPC models) and integrated models, for each cardiometabolic trait, rather than selecting the single best-performing PGS (based on max R2), all PGS were retained for subsequent predictive modelling analyses and elastic net regression was utilised to simultaneously select and weight predictors [40, 41]. Genetically inferred ancestry was included in prediction models to account for population stratification and potentially improve prediction [42]. To reduce overfitting, ancestry was determined by fitting data to the 1 KG Phase 3 projected principal components (PPCs) of population structure and not to AWI-Gen sample PCs.
Non-genetic models included ten conventional risk factors selected based on data availability in AWI-Gen and their known association with CVDs. Integrated models included genetic (PGS and PPCs) and non-genetic factors. No data were available for diet-related variables for the Soweto study site for men and women, so this site was excluded from prediction modelling analyses.
Statistical analysisAll analyses were performed using PLINK v1.9 (https://www.cog-genomics.org/plink/1.9/) [43], and R version 3.4.4 (http://www.r-project.org/) [44] unless specified otherwise. Data are presented as percentages (%) or mean ± SD. Associations between non-genetic factors and PGS and CVD-associated outcomes were assessed by logistic regression with the adjustment of PCs, sex, age, and age squared as described previously [15].
Ethics statementThis study was approved by the Human Research Ethics Committee (Medical) of the University of the Witwatersrand (Wits)(protocol number M210355) as a substudy of the AWI-Gen project (protocol number M170880).
Comments (0)