
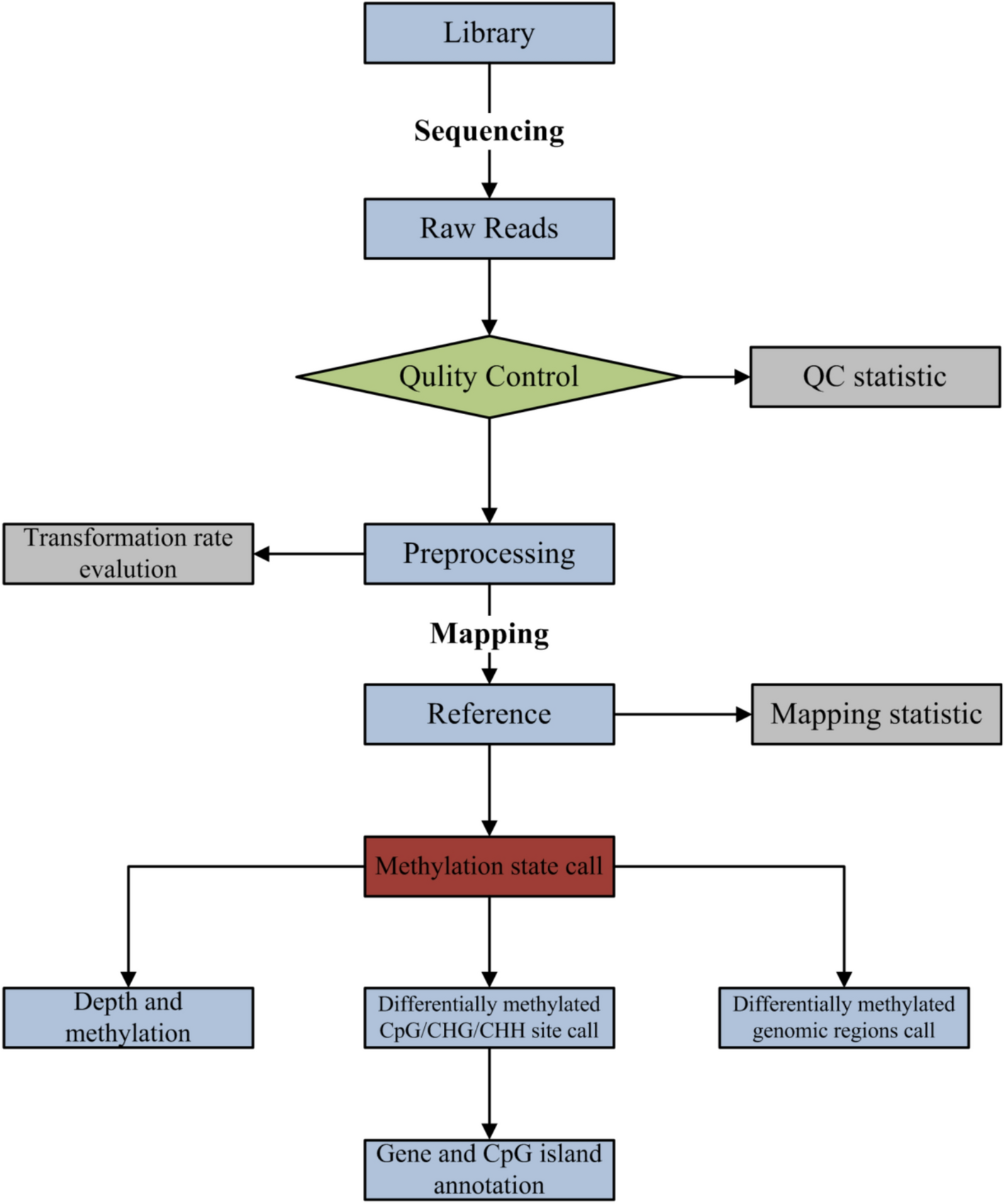
Remember me





Using various machine learning techniques, we developed a risk score model based on 12 genes associated with cell death. Figure 1 displays the C-index of several risk models associated with various algorithms in the validation sets GSE30219, and the training sets TCGA-LUAD, GSE31210, and GSE72094. Figure 1A shows that the model constructed with the coxboost rsf approach performed well predictively in the sets used for training and validation, with C-index values of 0.93, 0.67, 0.68, and 0.64, respectively. The CoxBoost RSF algorithm was selected for our risk score model due to its strengths in survival analysis. CoxBoost enhances the Cox model by addressing high-dimensional data, a common feature in genomic studies, through boosting techniques that improve prediction accuracy. The RSF component adds ensemble learning, capturing complex gene interactions and enhancing model generalizability. This combination is ideal for analyzing the intricate data from 12 cell death-associated genes, offering a robust method for patient stratification based on genetic profiles, as evidenced by strong predictive performance across our datasets [29]. Kaplan–Meier curves were constructed to analyze the prognostic differences between low-risk and high-risk groups for the training and validation sets (Fig. 1B). Overall training and validation sets, the prognosis for the low-risk group was significantly better than that of the high-risk group (p < 0.05). At 1, 3, and 5 years, robust AUC values were also observed in the risk score model for the training and validation sets (Fig. 1C). This was especially clear in the training set, which showed excellent performance prediction at 1, 3, and 5 years, respectively, with AUC values of 0.955, 0.980, and 0.962. Finally, based on the box boost of the method, 19 genes were included in the risk score model's creation (Figure S1A). Functional enrichment analysis shows the main signaling pathways the genes above enrich (Figure S1B). Figure S1C displays the inter-gene correlations, among which KRT8 and KRT18 exhibit a significant positive correlation.
Fig. 1
Performance of the 12-gene cell death-associated risk score model. A Comparison of the C-index across multiple risk score models based on various machine learning algorithms in the training set (TCGA-LUAD) and validation sets (GSE30219, GSE31210, and GSE72094). B Kaplan–Meier survival curves illustrating the significant prognostic differences between the high-risk and low-risk groups in both training and validation sets (p < 0.05). C Time-dependent ROC curves assessing the risk score model's accuracy at 1, 3, and 5 years in the training and validation sets, demonstrating strong AUC values
3.2 Functional difference analysis between high and low-risk groupsThe volcano graphic in Fig. 2A shows how differentially expressed genes, or DEGs, manifest in groups of high and low risk. Genes including SLC6A4, MYOC, RS1, and AGER were significantly downregulated, whereas KRT81, PADI1, KRT16, and so on were highly upregulated. The PPI network in Fig. 2B illustrates the main functions of proteins related to DEGs. It can be observed that DEGs are highly gathered in the functions of intrinsic components of the presynaptic membrane, spliceosomal snRNP complex, and myofibril, suggesting that DEGs might play key roles in these functions (Fig. 2D). A second KEGG analysis identified DEGs as being substantially abundant in the following processes: protein digestion and absorption, thiamine metabolism, malaria, and ECM-receptor interaction (Fig. 2C). Figure 2D of the GSVA analysis demonstrated that DEGs in the high-risk group were downregulated in the pathways of BILE ACID METABOLISM and FATTY ACID METABOLISM but raised in GLYCOLYSIS, E2F TARGETS, and G2M CHECKPOINT. Signaling pathway enrichment differences between the low- and high-risk groups, as indicated by the GO analysis results (Figure S2A). In high-risk individuals, there may be a strong association between increased tumor growth and poor prognosis, as shown in (Figures S2B-C). The results suggest that the high-risk group demonstrates significantly elevated scores compared to the low-risk group regarding homologous recombination deficiency (HRD), RNA stemness, DNAss, and DMP stemness (p < 0.05) (Fig. 2E).
Fig. 2
Differentially expressed genes (DEGs) and pathway enrichment analysis between high-risk and low-risk groups. A Volcano plot showing the DEGs between high- and low-risk groups. Genes such as SLC6A4, MYOC, RS1, and AGER were significantly downregulated, while KRT81, PADI1, and KRT16 were upregulated in the high-risk group. B Protein–protein interaction (PPI) network highlighting the key biological functions of DEGs, including components of the presynaptic membrane, spliceosomal snRNP complex, and myofibrils. C KEGG pathway enrichment analysis of DEGs revealing significant enrichment in pathways such as protein digestion and absorption, thiamine metabolism, malaria, and ECM-receptor interaction. D Gene Set Variation Analysis (GSVA) showing that pathways such as BILE ACID METABOLISM and FATTY ACID METABOLISM were downregulated, while pathways like GLYCOLYSIS, E2F TARGETS, and G2M CHECKPOINT were upregulated in the high-risk group. E Boxplots showing significantly higher homologous recombination deficiency (HRD), RNA stemness, DNAss, and DMP stemness scores in the high-risk group compared to the low-risk group (p < 0.05)
3.3 Immune infiltration analysis between high and low-risk groupsIn addition, we compared immune cell infiltration between low-risk and high-risk groups to identify the cause of prognostic differences between these groups. Figure 3A displays a heatmap showing the differences in expression levels between high-risk and low-risk groups regarding immune cell infiltration, stromal cell infiltration, and MeTIL score. Contrasting the groups at high and low risk, Fig. 3B and Figure S4A show that the former had significantly higher CAF infiltration and exclusion levels (p < 0.05), while the latter had much lower dysfunction levels (p < 0.05). Similarly, Fig. 3C illustrates how the high-risk group responded to the release of cancer cell antigen by having notably larger levels of Th1 cell, CD8 T cell, macrophage, eosinophil, NK, and basophil recruitment than the low-risk group did. The TIMER, CIBERSORT, QUANTISEQ, MCP-counter, x-cell, and EPIC algorithms are used in Figures S3A and S4A to show how immune cell infiltration differs in high- and low-risk groups. A heatmap in Figure S3B shows the expression differences of immune response regulatory components across high and low-risk groups, including chemokines, chemokine receptors, MHC, immune inhibitors, and immunostimulators. Additionally, as shown in Figure S4C, we examined the relationship between immune-related processes and risk score. In light of these findings, the efficacy of immunological therapy varied between high-risk and low-risk populations. The high-risk group's TIDE score was significantly higher than the low-risk group's (Figure S4B) score, suggesting a higher chance of immunotherapy benefit for the latter. The study also showed a substantial association between the TIDE score and the effectiveness of immunotherapy. As seen in Fig. 3D, the high-risk group's immunotherapy response rate (27%) was noticeably lower than that of the low-risk group (46%).
Fig. 3
Immune cell infiltration and immunotherapy response analysis in high-risk and low-risk groups. A Heatmap depicting differences in immune cell and stromal infiltration as well as the MeTIL score between high-risk and low-risk groups. B Boxplot comparing cancer-associated fibroblast (CAF) infiltration, exclusion levels, and dysfunction levels between the two groups, revealing significantly higher CAF infiltration and exclusion in the high-risk group and lower dysfunction levels in the low-risk group (p < 0.05). C Bar graph illustrating enhanced recruitment of immune cells such as Th1, CD8 T cells, macrophages, eosinophils, NK cells, and basophils in the high-risk group compared to the low-risk group. D Immunotherapy response rates in the high- and low-risk groups, showing a higher response rate (46%) in the low-risk group compared to the high-risk group (27%)
3.4 Mutation analysis between high and low-risk groupsThe top 20 genes for both Low-risk and high-risk groups are shown in Fig. 4A, B, together with the sorts of mutations that occur in each gene. TP53, TTN, and MUC16 showed the highest incidence of mutations in the low-risk group, according to an analysis of the mutation conditions across the groups. Three genes: TP53, TTN, and CSMD3. Missense mutations made up the majority of the mutations in the high-risk group. Next, we examined exclusivity relationships and co-mutations for the top 20 genes exhibiting the highest mutation frequencies in the high- and low-risk groups (Fig. 4C, D). Results revealed that most genes displayed co-mutation interactions, including MUC16 and NPAP1, TP53 and PCLO, etc., while TP53 and STK11, TP53 and KEAP1, TP53 and KRAS, and KRAS and PCLO established an exclusivity link within the low-risk group. The high-risk group had a strong exclusive relationship between TP53 and KRAS, while most genes showed significant co-mutation relationships, such as TTN and SI, CSMD3 and CDH10, USH2A and TNR, etc. The FNBP4, PPM1B, and PHF2 mutant genes in the low-risk group showed higher mutation frequencies than those in the high-risk group, according to a comparison of the mutated genes in the two groups. This finding suggests that these genes may function as tumor suppressors, preventing the growth and spread of tumors. (Fig. 4E).
Fig. 4
Mutation analysis of high-risk and low-risk groups. A, B Bar plots depicting the top 20 mutated genes in low-risk and high-risk groups, respectively. In the low-risk group. C, D Co-occurrence and exclusivity analysis of the top 20 genes revealed strong co-mutation interactions in both risk groups. E Comparison of the mutation frequencies for FNBP4, PPM1B, and PHF2 genes, showing higher mutation rates in the low-risk group, suggesting their potential tumor-suppressor role
3.5 Drug sensitivity analysis between high and low-risk groupsFigure 5A's box plots show how the IC50 values of several drugs differ for those at high risk compared to individuals at low risk. The findings indicated that under the backgrounds of CCT007093, Nutlin 3a, EHT 1864, MK2206, GW 441756, Erlotinib, VX702, PD 0332991, Roscovitine, AS601245, due to a lower IC50 value in the high-risk category, these drugs may have a greater impact on those at low risk. However, under the backgrounds of RO3306, BI2526, JNK inhibitor, GW 843682X, A 443654, Vinblastine, BID 1870, VX680, SL0101, etc., The IC50 value of the high-risk group was lower than that of the low-risk group, suggesting that they might be more susceptible to these drugs.
Fig. 5
Drug Sensitivity Analysis of High-risk and Low-risk Groups. Box plot showing the IC50 values of different drugs in high-risk and low-risk groups
3.6 Expression and function analysis of KRT18 in LUADWe selected KRT18, the gene that, while the risk score model was being created, had the highest weight coefficient for the subsequent study. In GSE19188, GSE31210, GSE68571, and TCGA-LUAD, KRT18 expression in the tumor was considerably higher (p < 0.05) than in the normal tissue, as shown in Fig. 6A. Similarly, KRT18 expression levels in tumor tissues with varying T-stages differed significantly (p < 0.05) in GSE13213 and TCGA-LUAD. (Fig. 6B). Next, the link between KRT18 and the COX regression analysis was examined and the prognosis of lung cancer further. Figure 6C's forest plot illustrates the strong correlation between high KRT18 expression and the shortened OS in GSE31210, GSE68571, GSE30219, GSE26939, and TCGA-LUAD; high KRT18 expression and shortened DFS in GSE14814, GSE30219; high KRT18 expression and shortened RFS in GSE31210; high KRT18 expression and shortened DSS and PFS in TCGA-LUAD. These findings point to KRT18 as a significant determinant of lung cancer prognosis. The GSEA functional analysis shows that KRT18 is highly enriched in pathways such as DNA unwinding involved in DNA replication, mitochondrial translation, and mitochondrial gene expression, as well as in hallmarks such as E2F targets, MYc targets, and G2m checkpoint (Figs. 6D, E). Next, we analyzed the correlation between KRT18 and mutations and copy numbers of other genes. The heatmap displayed in Fig. 6F shows that the group with high and low KRT18 expression had significantly different (p < 0.05) mutation levels of MUC16, USH2A, KRAS, and ANK. The groups with high levels of KRT18 expression are compared to those with low levels in Fig. 6F, where copy number amplification or deletions are also displayed. In order to study the effects of KRT18 on LUAD drug resistance, researchers calculated the correlation between KRT18 and IC50 values of various drugs in LUAD across different cohorts, as shown in the heatmap in Fig. 6G.
Fig. 6
Expression and functional analysis of KRT18 in lung adenocarcinoma (LUAD). A Boxplots showing KRT18 expression levels in tumor and normal tissues across multiple datasets (GSE19188, GSE31210, GSE68571, TCGA-LUAD), demonstrating significantly higher expression in tumor tissues (p < 0.05). B KRT18 expression levels in different T-stages in GSE13213 and TCGA-LUAD, showing significant differences (p < 0.05). C Forest plot illustrating the relationship between high KRT18 expression and overall survival (OS), disease-free survival (DFS), recurrence-free survival (RFS), and other prognostic indicators in various datasets, revealing its association with poor prognosis. D, E GSEA functional analysis indicating KRT18 enrichment in pathways related to DNA replication, mitochondrial translation, and key hallmark processes such as E2F targets, MYC targets, and G2M checkpoint. F Heatmap displaying significant differences in mutation levels of MUC16, USH2A, KRAS, and ANK between high and low KRT18 expression groups. Copy number amplification and deletions are also shown. G Heatmap showing the correlation between KRT18 expression and drug sensitivity (IC50 values) in LUAD across various cohorts
3.7 KRT18 expression and functional analysis across various Cancer typesWe then compared KRT18 expression levels in tumors and healthy tissue and in various cancer types. As shown in Fig. 7A, KRT18 is significantly overexpressed in GBM, UCEC, BRCA, CESC, and OV tumor tissues, indicating KRT18's potential to promote the development of these cancers. In contrast, KRT18 is significantly under-expressed in GBMLGG, LGG, WT, SKCM, and ALL tumor tissues, suggesting that KRT18 may inhibit the progression of these cancers (p < 0.05). Epigenetic modifications can generally be categorized into three main aspects of gene regulation: writers, readers, and erasers. The writer-reader-eraser system enables precise cell regulation. Using the m1A, m5C, and m6A changes as a reference, we examined the connection between KRT18 expression and the genes associated with writers, readers, and erasers (Fig. 7B).
Fig. 7
KRT18 expression and functional analysis across multiple cancer types. A Boxplots comparing KRT18 expression between tumor and normal tissues in various cancer types, showing significant overexpression in GBM, UCEC, BRCA, CESC, and OV, and underexpression in GBMLGG, LGG, WT, SKCM, and ALL (p < 0.05). B Heatmap illustrating the correlation between KRT18 expression and epigenetic regulators (writers, readers, erasers) associated with m1A, m5C, and m6A modifications. C Heatmap showing the relationship between KRT18 expression and immune cell infiltration across various cancer types, highlighting its impact on immune recruitment in different tumors. D Heatmap analyzing the correlation between KRT18 expression and immune stimulatory and inhibitory molecules across multiple cancer types
In addition, we looked at the connection between KRT18 and the level of immune cell infiltration in various cancer types (Fig. 7C). KRT18 inhibits LUAD, PPRAD, CHOL, TGCT, CD8 T cells, CD4 T cells, DCs, and macrophages from infiltrating different areas. However, UCS, PCPG, LIHC, LGG, GBM, and KRT18 recruit B cells, macrophages, CD4 T cells, CD8 T cells, neutrophils, and DCs. We examined the relationship between immune stimulatory and inhibitory chemicals and KRT18 expression levels in various cancer types (Fig. 7D).
Comments (0)