
Remember me
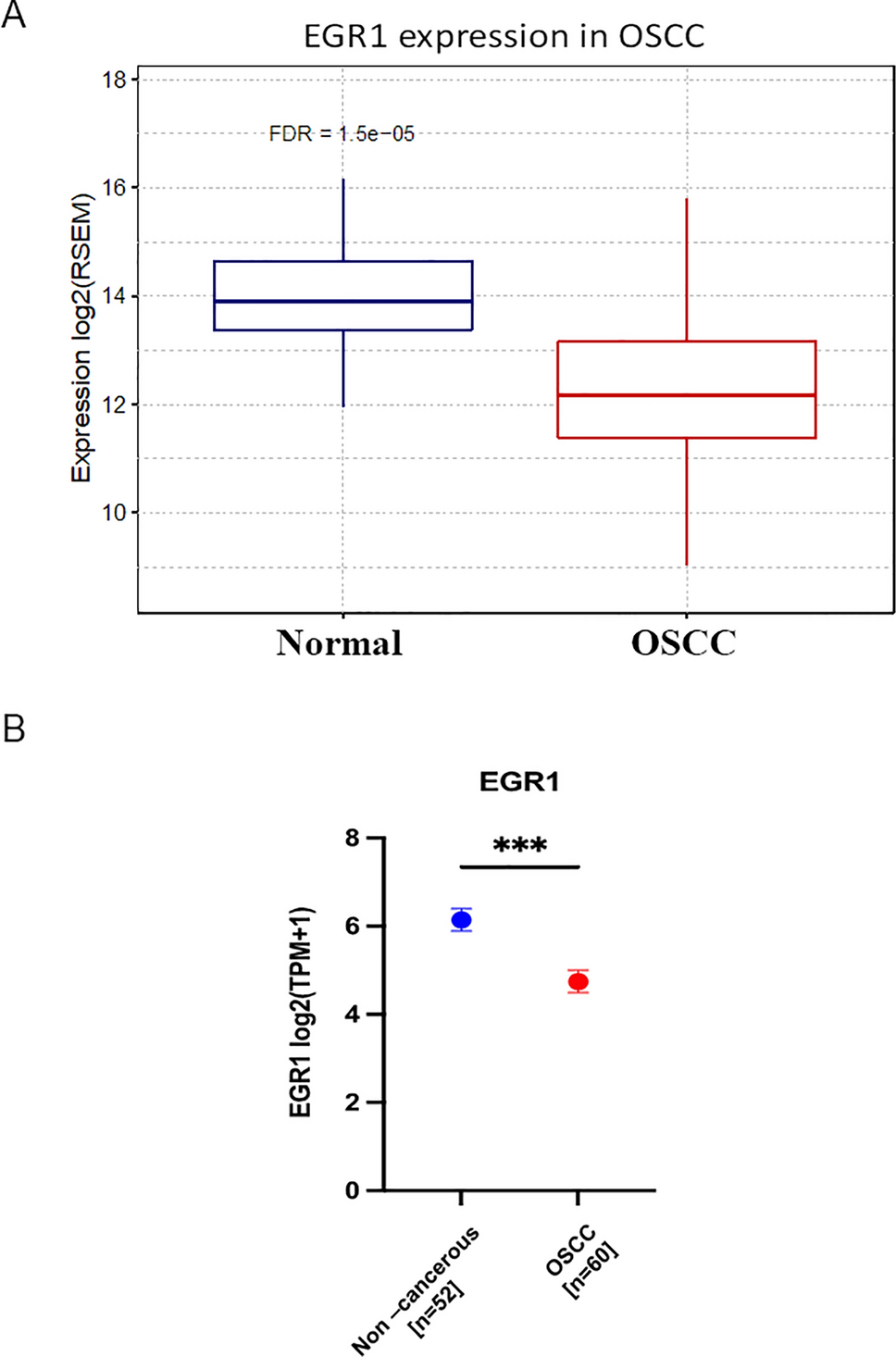





The “limma” package was used to identify DEGs in the GEO dataset GSE56363 based on the screening criteria of p-values of < 0.05 and log2FC values of 1.5. A total of 152 DEGs were identified between patients with CESC with a CR and those with a non-CR to radiotherapy. Of the 152 DEGs, 13 genes were downregulated, whereas 139 genes were upregulated. Figure 1A and B show a volcano plot and heatmap demonstrating DEGs, respectively. Online Resource Table 1 shows all identified DEGs.
Fig. 1
Volcano plot and heatmap of differentially expressed genes (DGEs) between patients with CESC with a complete response (CR) and those with a non-complete response (non-CR) to radiotherapy A Volcano plot of DEGs. Grey represents non-significant differentially expressed genes, green represents genes with log2FC values of > 1.5 or < − 1.5 but p-values of > 0.05, blue represents genes with p-values of < 0.05 but log2FC values of > − 1.5 and < 1.5, and red represents genes with log2FC values of > 1.5 or < − 1.5 and p-values of < 0.05. B Heatmap of DEGs between patients with CESC with a CR (aquamarine) and those with a non-CR (pink-orange) to radiotherapy. Blue represents a low expression level, whereas red represents a high expression level
GO and KEGG enrichment analyses were performed to investigate the biological functions and pathways of DEGs. The results indicated that the DEGs were primarily enriched in pathways related to the extracellular matrix, components of the extracellular matrix that provide elasticity, and heparin binding (Online Resource Fig. 1). The results of PPI analysis showed that the interactions among DEGs were both intricate and compact (Online Resource Fig. 2).
3.2 Random survival forest analysis of DEGs in the TCGA-CESC cohort and identification of six hub genesRandom survival forest analysis revealed seven genes associated with the prognosis of CESC, namely, SELP, PIM2, CCL19, SDS, PRR3, NRP1, and SF3A2 (Fig. 2A and B). Kaplan–Meier analysis showed that all genes except PRR3 were significantly associated with survival (Fig. 2D–I). In particular, higher expression of SELP, PIM2, CCL19, SDS, and SF3A2 (p < 0.05 for all) and lower expression of NRP1 (p = 0.041) were significantly associated with longer OS. The expression of six hub genes was significantly higher in the CR group than in the non-CR group in the GSE56363 dataset (all p < 0.05, Fig. 2C).
Fig. 2
Random survival forest analysis of DEGs in the TCGA-CESC cohort A Random survival forest analysis of DEGs. B Seven final genes with variable relative importance values of ≥ 0.4. C The expression of six hub genes was significantly higher in the CR group than in the non-CR group (*p-values of < 0.05, **p-values of < 0.01, and ***p-values of < 0.001). D Kaplan–Meier analysis showed that higher expression of SELP, PIM2 E, CCL19 F, SDS G, and SF3A2 I was significantly associated with longer overall survival, whereas higher expression of NRP1 H was associated with shorter OS (p < 0.05 for all)
3.3 Relationship between hub genes and tumor-infiltrating immune cellsFigure 3A and B show the distribution of 22 types of tumor-infiltrating immune cells in each sample and the relationship among these cells, respectively. As shown in Fig. 3C, the proportion of M0 macrophages, follicular helper T cells, and M1 macrophages was significantly higher in patients with CESC than in healthy individuals. The expression of PIM2, SDS, and SELP was significantly correlated with the abundance of tumor-infiltrating immune cells (Fig. 3D). In particular, PIM2 expression was positively correlated with the proportion of plasma cells, CD8 T cells, activated memory CD4 T cells and negatively correlated with the proportion of resting memory CD4 T cells and activated dendritic cells. SDS expression was positively correlated with the proportion of activated memory CD4 T cells and M1 macrophages and negatively correlated with the proportion of resting memory CD4 T cells and activated mast cells. SELP expression was positively correlated with the proportion of resting mast cells (Fig. 3D).
Fig. 3
Relationship between hub genes and tumor-infiltrating immune cells in the TCGA-CESC cohort A Proportion of immune cells in the control and CESC groups. Red, control group; dark red, CESC group. B Interaction analysis of 22 types of immune cells in CESC (Pearson correlation coefficients are shown for significant correlations).C Comparison of immune cells between the control and CESC groups (*p-values of < 0.05, **p-values of < 0.01, and ns represents p-values of > 0.05 between the two groups). D Bubble map demonstrating the correlation between hub genes and tumor-infiltrating immune cells (Pearson correlation coefficients are shown for significant correlations)
Furthermore, the relationship between hub genes and various immune-related genes, including chemokine-, immunosuppression-, MHC-, immunostimulation-, and receptor-related genes, was evaluated using the TISIDB database. As shown in Online Resource Fig. 3, the expression of the six hub genes was either positively or negatively correlated with that of various immune-related gens.
3.4 Response to chemotherapeutic drugs based on the expression of hub genesCommon chemotherapeutic drugs, including cisplatin, docetaxel, fluorouracil, and paclitaxel, were selected for drug sensitivity analysis. The expression of SELP, SDS, and CCL19 was significantly correlated with lower IC50 values of the aforementioned drugs, whereas the expression of NRP1 was significantly correlated with higher IC50 values of the drugs (p < 0.05 for all) (Online Resource Fig. 4). On the contrary, PIM2 did not affect the sensitivity to fluorouracil (p = 0.11), and SF3A2 did not affect the sensitivity to docetaxel (p = 0.33).
3.5 Relationship between the six hub genes and disease-related genesThe expression of numerous disease-related genes was significantly different between the control and CESC groups. These genes included RAD51, UHRF1, FANCI, BRCA2, PDGFRA, POLE, CDKN2A, ERBB3, IDH2, TGFBR2, HTATIP2, SOX5, SOX9, NRAS, FGFR3, SMAD4, HRAS, ATR, PTEN, PIK3R1, PTCH1, and ERBB2 (Fig. 4A). Pearson correlation analysis indicated a strong positive correlation between the expression of the six hub genes and that of the screened disease-related genes. As shown in Fig. 4B, the expression of SELP and NRP1 was positively correlated with that of PIK3R1, PDGFRA, SOX5, and TGFBR2; the expression of PIM2, SDS, and SF3A2 was positively correlated with that of ATR, IDH2, CDKN2A, POLE, and RAD51; and the expression of CCL19 was positively correlated with that of FGFR3 and PDGFRA.
Fig. 4
Relationship between hub genes and disease-related genes A Differences in the expression of multiple disease-related genes between the control and CESC groups (*p-values of < 0.05 and **p-values of < 0.01). B Bubble map demonstrating the correlation between the six hub genes and disease-related genes (Pearson correlation coefficients are shown for significant correlations)
3.6 Signaling pathways associated with the six hub genesFurthermore, we identified specific signal transduction pathways associated with the six hub genes and evaluated the potential effects of the genes on the identified pathways during CESC development. Figure 5A–C and Online resource Fig. 5A–C show enriched pathways identified through GSVA of the six hub genes. The results indicated that upregulated SELP was primarily enriched in pathways associated with abnormal vascular physiology and CD8 + α-β T cell differentiation. Upregulated PIM2 was primarily enriched in pathways associated with the regulation of cell death, stress-activated protein kinase signaling cascade, double-strand break sites, and PRP19 complex. Upregulated CCL19 was involved in the NF-kappa B pathway-induced kinase activity, whereas downregulated CCL19 was involved in the negative regulation of the cellular response to hypoxia. Upregulated SDS was primarily enriched in pathways associated with the regulation of mast cell activation involved in immune response, regulation of mononuclear cell migration, leukocyte activation involved in the inflammatory response, and granzyme-mediated programmed cell death signaling. Upregulated NRP1 was associated with protein localization to the cell leading edge. Downregulated SF3A2 was primarily enriched in pathways associated with the regulation of cilium-dependent cell motility, forelimb morphogenesis, and ERBB4 signaling, whereas upregulated SF3A2 was associated with the regulation of cysteine endopeptidase activity involved in the apoptotic signaling pathway.
Fig. 5
GSVA and GSEA of the six hub genes A GSVA of SELP. B GSVA of PIM2. C GSVA of SDS. D GSEA of SELP. E GSEA of PIM2. F GSEA of SDS
Furthermore, GSEA showed that upregulated SELP, PIM2, CCL19, SDS, and SF3A2 were enriched in pathways associated with allograft rejection and graft-versus-host disease (Fig. 5D–F and Online resource Fig. 5D–F).
3.7 Establishment of a nomogram and calibration curve to predict the OS of patients with CESCCox analysis revealed that clinical variables and the expression of the six hub genes played distinct roles in the CESC scoring process. As the total score of these characteristics increases, the probability of 1-, 3-, and 5-year OS decreases (Fig. 6A). The calibration curve showed that the predicted survival rates were in good agreement with the actual outcomes (Fig. 6B).
Fig. 6
Nomogram for predicting the overall survival of patients with CESC A A nomogram was constructed based on the expression of the six hub genes and clinical factors. B Calibration curve of the nomogram for predicting 1-, 3-, and 5-year OS in the TCGA-CESC cohort
3.8 Regulatory network analysis and ceRNA network analysis of hub genesThe six hub genes, namely, SELP, PIM2, CCL19, SDS, NRP1, and SF3A2, were found to be regulated by various TFs. To investigate the regulatory mechanisms of these genes, motif enrichment analysis was performed on predicted TFs (Fig. 7A). The jaspar_MA0067.1 motif had the highest NES at 7.25 (Fig. 7B), followed by cisbp_M189 with an NES of 7.23 and predrem_nrMotif423 with an NES of 7.15 (Fig. 7C and D). NRP1, PIM2, and SELP were enriched in jaspar_MA0067.1, with no upstream TFs. A portion of the enriched motifs and their respective TFs for the hub genes are shown in Fig. 7E.
Fig. 7
Regulatory network analysis of hub genes A Enrichment analysis of transcription factors of hub genes using the “RciTarget” package. B The jaspar_MA0067.1 motif. C The cisbp_M1895 motif. D The predrem_nrMotif423 motif. E Enriched motifs and corresponding TFs of hub genes
A total of 331 CESC-related miRNAs were identified using the HMDD database, and a total of 1140 mRNA–miRNA pairs for the six hub genes were identified using the miRWalk database. Only mRNA–miRNA pairs involving CESC-related miRNAs were retained, resulting in the inclusion of 15 miRNAs and 4 mRNAs (Fig. 8A). Furthermore, a total of 301 miRNA–lncRNA pairs, involving 9 miRNAs and 212 lncRNAs, were identified using the ENCORI database. The results showed that SELP was regulated by hsa-miR-107 and hsa-miR-665; PIM2 was regulated by hsa-miR-665, hsa-miR-4428, and hsa-miR-320c; CCL19 was regulated by hsa-miR-326 and hsa-miR-543; and NRP1 was regulated by hsa-miR-449a, hsa-miR-613, and hsa-miR-1270. Complex ceRNA networks involving miRNAs and lncRNAs were constructed using the Cytoscape software (Fig. 8B).
Fig. 8
ceRNA network analysis of hub genes A A total of 366 miRNAs related to CESC were obtained from HMDD, and 1140 miRNAs related to the six hub genes were obtained from the miRWalk database. B ceRNA networks involving mRNAs, miRNAs, and lncRNAs
3.9 GWAS analysis of hub genesBecause the Gene Atlas database does not contain information on cervical cancer, we used the C51-C58 malignant neoplasms of female genital organs as a trait for GWAS analysis in this study (Fig. 9A, B). The pathogenic single nucleotide polymorphisms (SNPs) corresponding to SELP, CCL19, SDS, NRP1, and SF3A2 genes are shown in Fig. 9C–G. The results showed that the pathogenic SNPs of SELP, CCL19, SDS, NRP1, and SF3A2 were located on chromosomes 1, 9, 12, 10, and 19, respectively. Data on PIM2 were not found in the Gene Atlas database.
Fig. 9
Results of GWAS analysis A Q-Q plot of GWAS analysis. B Manhattan plot of the GWAS analysis. C The pathogenic region of SELP was located on chromosome 1. D The pathogenic region of CCL19 was located on chromosome 9. E The pathogenic region of SDS was located on chromosome 12. F The pathogenic region of NRP1 was located on chromosome 10. G The pathogenic region of SF3A2 was located on chromosome 19
3.10 Validation of the expression of the six hub genes in clinical samplesFigure 10 shows the results of immunohistochemical staining for SELP, PIM2, CCL19, SDS, NRP1, and SF3A2 in cervical tissues obtained from the HPA database. SELP was not expressed in normal cervical tissues, and its expression was either undetected or at a moderate level in cervical cancer tissues. PIM2 was not expressed in normal cervical tissues, and its expression was either undetected or low in cervical cancer tissues. CCL19 was expressed in neither normal cervical tissues nor cervical cancer tissues. SDS showed moderate expression in normal cervical tissues, whereas its expression was either undetected or at a low-to-moderate level in cervical cancer tissues. NRP1 exhibited low expression in normal cervical tissues, and its expression was either undetected or at a low-to-moderate level in cervical cancer tissues. SF3A2 showed moderate expression in normal cervical tissues and moderate-to-high expression in some parts of cervical cancer tissues. The varying expression patterns of the hub genes may account for the innate biological differences and differences in the response to radiotherapy among patients with CESC.
Fig. 10
Protein expression of SELP, PIM2, CCL19, SDS, NRP1, and SF3A2 in normal cervical tissues and cervical cancer tissues from the HPA database
Comments (0)