
Remember me
Quantitative in vitro characterisation of the metabolism and inhibition properties of VRC has been previously performed by Schulz et al. [17]. The Michaelis–Menten kinetics of NO formation by CYP2C19, CYP2C9 and CYP3A4 were determined in incubations of human recombinant CYP enzymes and in liver and intestine microsomes. In addition, the inhibitory potential, and the mechanism of inhibition of VRC, NO and OHVRC, were evaluated by their effects on CYP marker reactions. Overall, these quantitative in vitro assays provided a basis for the a priori PBPK modelling of VRC and its metabolites.
Individual clinical data, comprising plasma concentration-time profiles of VRC and its two metabolites (NO and OHVRC), were obtained from three clinical studies [13, 14, 30] conducted in healthy adults at the University Hospital Heidelberg. The studies primarily investigated intravenous subtherapeutic and therapeutic dosing regimens, covering a broad dose range (50–400 mg) of single doses (SDs), and allowed stratification based on CYP2C19 genotype. Specifically, doses of 50 mg infused over 2 h and 100 mg infused over 4 h were categorised as subtherapeutic, while 400 mg infused over 2 h was defined as a therapeutic dose. Moreover, urine samples were collected over a 24-h period in two of the three studies [13, 14], and the concentrations of VRC and both metabolites were measured. These concentrations, along with the recorded urine volumes, were used to calculate the amount of each analyte excreted unchanged in urine. In addition to the SD studies, the model development and evaluation also incorporated digitised mean VRC plasma concentrations from two additional clinical studies [37] that investigated intravenous multiple dosing (MD) regimens. One study (study A) reported individual maximum plasma concentration (Cmax) values for VRC on days 1, 5, 8 and 12, while the other (study B) provided individual minimum plasma concentration (Cmin) from the first to the tenth day of MD, and these data were digitised to further refine and validate the model. A detailed list of all clinical studies used is provided in Table 1.
Table 1 Clinical studies used for model development and evaluation2.2 Physiologically-Based Pharmacokinetic (PBPK) Model DevelopmentAn intravenous adult PBPK model was developed using in vitro data, in silico calculated and in vivo clinical data moving from a pure bottom-up approach to a middle-out approach. These approaches are discussed in the following paragraphs and the input parameters used are summarised in Tables 2 and 3. Figure 1 depicts the entire model development workflow along with the implemented metabolic pathways in Fig. 2 (right panel).
Table 2 Drug-dependent parameters of the voriconazole a priori PBPK modelTable 3 Drug-dependent parameters of the metabolites (voriconazole N-oxide and hydroxyvoriconazole) a priori PBPK modelsFig. 1
Overview of the PBPK model development workflow for voriconazole and its metabolites, illustrating the input/data sources (inside boxes, left side) and the achieved steps (inside boxes, right side). ADME absorption, distribution, metabolism, excretion, CYP cytochrome P450, gXM genotype-predicted phenotype, H hypothesis, IM intermediate metaboliser, i.v. intravenous, MM Michaelis–Menten, NM normal metaboliser, NO voriconazole N-oxide, OHVRC hydroxyvoriconazole, PBPK physiologically-based pharmacokinetic, PM poor metaboliser, RM rapid metaboliser, VRC voriconazole
Fig. 2
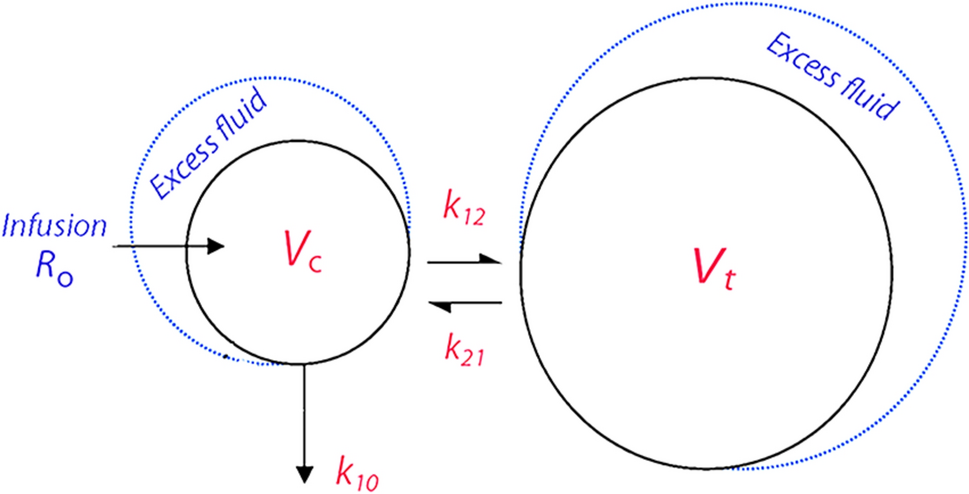
Whole-body intravenous physiologically-based pharmacokinetic model for voriconazole and its metabolites (left) and schematic representation of voriconazole elimination pathways (right). Voriconazole is metabolised by CYP2C19, CYP3A4 and CYP2C9 into voriconazole N-oxide, with subsequent elimination via a nonspecific hepatic route and excreted via renal clearance. The remainder of voriconazole absorbed is transformed via CYP3A4 into hydroxyvoriconazole, which is further metabolised and excreted via renal clearance. The parent compound and both metabolites concomitantly inhibit CYP2C19, CYP3A4 and CYP2C9. CYP cytochrome P450
2.2.1 Bottom-Up ApproachInitially, a pure bottom-up approach was adopted to develop an intravenous a priori PBPK model of the parent compound (VRC), based on literature-derived physicochemical properties and information regarding its distribution, metabolism, and excretion processes (Fig. 1, first blue box). Partitioning into tissues was modelled by the Berezhkovskiy tissue distribution model as implemented in PK-Sim® [38]. The elimination processes involved hepatic metabolism and renal excretion: VRC was assumed to be metabolised via CYP2C19, CYP3A4 and CYP2C9, with a renal CL process implemented to account for the minor excretion of unchanged VRC via the kidneys. Initial values for renal CL were based on literature data using the ratio of the amount excreted in urine to the area under the concentration–time curve (AUC) over 24 h [12, 39]. The relative tissue-specific expression of enzymes was taken from the PK-Sim® expression database based on high-sensitive real-time reverse transcription-polymerase chain reaction (RT-PCR) profiles [40], together with a reference concentration of 0.76 μmol of CYP2C19, 4.32 μmol of CYP3A4 and 3.84 μmol of CYP2C9 per litre of liver tissue [41] that were incorporated in the model.
Subsequently, the a priori parent-model was expanded to include the two metabolites, NO and OHVRC, guided by an exhaustive literature search for their physicochemical properties and distribution, metabolism, and excretion processes (Fig. 1, second blue box). NO was assumed to be formed by the three included CYP enzymes [17], while OHVRC was assumed to be formed via CYP3A4 only [42]. Due to the lack of definitive information regarding the elimination processes of both metabolites, we initially assumed that NO was eliminated through hepatic and renal CL processes. The total hepatic CL of NO was described as a first-order process in the liver derived from plasma CL, back-calculated from a published nonlinear mixed-effects (NLME) model [43], as input value. For OHVRC, we assumed only renal elimination, based on findings from literature suggesting that hydroxy metabolites are more commonly excreted through the kidneys [14] (see Fig. 2, right panel). The renal CL processes for both metabolites were implemented in a similar manner to the parent, with initial values based on literature data, calculated as the amount excreted in urine divided by the corresponding AUC values [12].
Furthermore, to capture the complex interactions, specifically the (auto)-inhibition of VRC and the inhibition mechanisms of NO and OHVRC on each CYP enzyme, equations from multiple inhibition analyses [44] were integrated into the coupled a priori parent-metabolite PBPK model using MoBi®. The reversible competitive inhibition of CYP2C19 and CYP2C9 by VRC and its metabolites was modelled using Eqs. 1 and 2, while the reversible noncompetitive inhibition of CYP3A4 was modelled using Eqs. 1 and 3, based on the respective in vitro data [17].
$$v = \frac \times C_ }} + C_ }}$$
(1)
$$K_ = K_ \left( }} }} + \frac }} }} + \frac }} }}} \right)$$
(2)
$$V_ = \frac }} }} }} + \frac }} }} + \frac }} }}} \right)}}$$
(3)
In Eqs. 1–3, \(v\) is the velocity of the reaction (rate of metabolism via CYP enzyme), \(_\) is the unbound concentration of substrate (VRC), \(_\) is the apparent Michaelis–Menten constant representing half the maximum reaction velocity in the presence of an inhibitor, \(_\) is the Michaelis–Menten constant representing half the maximum reaction velocity (measured in vitro), \(_\) is the apparent maximum reaction velocity in presence of an inhibitor, \(_\) is the maximum reaction velocity (measured in vitro), and \(_\) is the reversible inhibition constant.
Virtual individuals and populations [45] were created to represent each clinical study included in our modelling workflow based on their respective median data for age, weight, height, and body mass index. The relative expression of enzymes of interest in various organs was determined using the open system pharmacology (OSP) human gene expression database [40]. Different CYP2C19 genotype-predicted phenotypes were incorporated into the model using varying CYP2C19 concentrations (Fig. 1, third blue box). Initially, a virtual population of 1000 individuals was generated based on the demographic characteristics of the clinical studies. For characterising the distribution of hepatic concentrations of CYP2C19 within the simulated population, a log-normal distribution was assumed around the reference CYP2C19 normal metaboliser (NM) concentration value of 0.76 µmol/L with a geometric standard deviation (GeoSD) of 1.79 derived from the OSP human gene expression database [46]. Based on the match between the frequencies of CYP2C19 genotype-predicted phenotype (gXM) in our clinical database and those reported by the Clinical Pharmacogenetics Implementation Consortium (CPIC) for the Caucasian population [33] (Fig. S1 in the electronic supplementary martial [ESM]), four distinct subpopulations, each of 1000 individuals, were constructed. The variability in the CYP2C19 expression of these subpopulations was incorporated into the model using a uniform distribution, constrained by minimum and maximum values determined by calculated percentiles of the reported frequencies of CYP2C19 gXM in the Caucasian population [33]. As a result, the CYP2C19 enzyme concentrations for PM (*2/*2, *2/*3, *3/*2), intermediate metaboliser (IM; *1/*2, *1/*3, *2/*17), NM (*1/*1), and rapid metaboliser (RM)/UM (*1/*17, *17/*17) phenotypes ranged from 0.001 to 0.24, 0.25 to 0.55, 0.56 to 0.95, and 0.96 to 3.55 μmol/L, respectively. Further details on the respective enzyme expression can be found in Table 4.
Table 4 System-dependent parameters and expression of relevant enzymes for DME2.2.2 Middle-Out ApproachDuring model development, a middle-out approach was employed, utilising clinical data to infer (1) model parameters that could not be substantiated by literature or in vitro analyses, as well as (2) influential parameters identified through local sensitivity analyses. Given the model’s complexity, a stepwise approach was adopted within a learn-confirm-predict paradigm to develop the a posteriori coupled parent-metabolite PBPK model.
First, CYP3A4-mediated metabolism was estimated based on data from CYP2C19 PM individuals, assuming that only CYP3A4 contributes to the formation of OHVRC and NO in this population (Fig. 1, first green box). Both the CYP2C19-mediated metabolism and renal CL processes were then estimated using plasma concentration-time profiles and the amount excreted in urine of single and multiple intravenous doses in CYP2C19 IM, NM and RM individuals (Fig. 1, second green box).
In subsequent estimation steps, it became clear that a consistent misprediction of the observed NO PK profiles persisted across different dosing regimens, even upon estimating influential parameters. Therefore, two hypotheses were generated in order to characterise the nonlinear elimination of NO (Fig. 1, third green box).
2.3 PBPK Model EvaluationThe parent-metabolite model was evaluated both visually and quantitatively. Predicted concentration-time profiles of VRC, NO and OHVRC were compared with observed data from the corresponding clinical studies. Additionally, predicted plasma concentration values for all studies were compared with their respective individual observed plasma concentrations using goodness-of-prediction (GOP) and goodness-of-fit (GOF) plots. Model performance was further assessed by comparing predicted values with observed values for the AUC from the time of drug administration to the time of the last concentration measurement (AUClast) and for Cmax. In both models, the predicted and observed AUClast were computed using a linear-up log-down trapezoidal method.
Quantitative metrics used to evaluate model performance included the root mean squared error (RMSE) for all predicted and observed plasma concentrations, as well as the average fold error (AFE) and absolute average fold error (AAFE) for all predicted and observed plasma concentrations and PK parameters. These were calculated using Eqs. 4–6.
$$RMSE = \sqrt ^ \left( - Predicted_ } \right)^ }$$
(4)
$$AFE = 10^ \quad with \quad x = \mathop \sum \limits_^ log_ \left( }} }}} \right)$$
(5)
$$AAFE = 10^ \quad with \quad x = \mathop \sum \limits_^ log_ \left| }} }}} \right)} \right| .$$
(6)
In Eqs. 4–6, \(_\) is the ith observed plasma concentration or AUClast or Cmax, \(_\) is the corresponding predicted plasma concentration or AUClast or Cmax, and n is the number of observed values (studies for PK parameters).
2.4 Sensitivity AnalysisLocal sensitivity analyses were conducted on both the a priori and a posteriori coupled parent-metabolite PBPK models to assess the impact of single parameter changes. These analyses allowed identifying which of the input parameters significantly influenced the model predictions of VRC, NO and OHVRC exposure. The parameters analysed included those derived from literature, associated with estimated parameters, or assumed to influence the predictions due to calculation methods in the model. The sensitivity of the model to an input parameter was quantified by the ratio of the relative change in the predicted PK metric (AUClast and Cmax) to the relative variation of the input parameter, as outlined in Eq. 7.
$$S = \frac} \times \frac$$
(7)
In Eq. 7, \(S\) is the sensitivity index of the PK metric to the evaluated model input parameter, \(\Delta PK \,metric\) is the change of the predicted PK metric, \(PK \,metric\) is the predicted PK metric with original model parameter value, \(\Delta p\) is the change of the assessed model parameter value, and \(p\) is the original model parameter value (initial input parameter value).
2.5 SoftwareDevelopment of the PBPK model, parameter estimation, simulations, and local sensitivity analyses were conducted using the open-source modelling software PK-Sim® and MoBi® (Open Systems Pharmacology Suite 11.2) [47, 48]. The parameter estimation routine employed the Monte Carlo algorithm, allowing a maximum of 10,000 iterations, and incorporated multiple identification options with randomised start values to ensure the model robustness. Individual PK profiles and amounts excreted in urine for each analyte were used in estimating the input model parameters identified from sensitivity analyses, with the goal of minimising the residual sum of squares (total error) between the simulated and observed data. Additionally, 95% confidence intervals (CIs) for the parameter estimates were derived using the Fisher information matrix. These intervals quantified the uncertainty associated with each parameter estimate, indicating the sensitivity of the total error to changes in parameter values. Published clinical study data were digitised using WebPlotDigitizer (version 4.2 [49]), according to best practices [50]. Model performance metrics and post-processing of simulations were executed in R 4.2.1 (The R Foundation for Statistical Computing, Vienna, Austria) with RStudio 2023.03.0+386 (RStudio Inc., Boston, MA, USA).
Comments (0)