
Remember me



Lipids play a pivotal role in cancer development and progression, with dysregulated lipid metabolism closely linked to key aspects of cancer biology, including initiation, growth, metastasis, and drug resistance [64]. Alterations in lipid metabolism confer survival advantages to cancer cells, such as enhanced resistance to chemotherapy and targeted therapies [65]. This metabolic reprogramming enables cancer cells to evade the cytotoxic effects of treatment, leading to increased resilience and complicating therapeutic outcomes [66]. Consequently, lipid dysregulation presents a significant challenge in cancer management, impairing the efficacy of conventional therapies.
Understanding the complex role lipids play in cancer—spanning initiation, progression, metastasis, and drug resistance—is critical for developing novel treatment approaches. Targeting lipid metabolism holds the potential to improve patient outcomes and overcome therapeutic resistance. Further research is essential to establish the intricate relationship between lipid dysregulation and cancer, leading to the establishment of new therapeutic targets and more effective interventions.
Altered membrane dynamics and oncogenic signalling drive cancer initiationIncreased lipid accumulation within the tumor microenvironment (TME) is closely associated with enhanced cancer invasion [67]. Dysregulated lipid metabolism alters the composition of cell membranes, impacting their fluidity, receptor distribution, and associated signaling pathways. These changes can promote uncontrolled cell growth, facilitating cancer initiation. Specific lipids, such as phosphoinositides and sphingolipids, are critical regulators of intracellular signaling, and their dysregulation can activate oncogenic pathways, further driving cancer initiation and progression [68].
FAs are a crucial energy source for various cellular processes, and they serve as the structural backbone of phospholipids and glycolipids in cell membranes [69]. There is a strong correlation between inflammation, metabolic syndrome, and increased consumption of saturated fatty acids (SFAs), particularly palmitate and stearate. However, polyunsaturated fatty acids (PUFAs) and other unsaturated fatty acids, such as oleate and linoleate, have been shown to mitigate or resolve inflammation [70].
Notably, palmitate, rather than glucose or insulin, has been identified as a key driver of the inflammatory and metabolic profile of adipose tissue macrophages (ATMs) in obese adipose tissue (AT) [71]. Palmitate triggers the release of inflammatory cytokines by macrophages through the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a central regulator of inflammation [72]. Additionally, both stearic and palmitic acids bind to Toll-like receptors (TLRs), activating the c-Jun N-terminal kinase (JNK) and NF-κB signaling pathways [73]. This process leads to increased levels of pro-inflammatory molecules, including monocyte chemoattractant protein-1 (MCP-1), interleukin-1 beta (IL-1β), and tumor necrosis factor-alpha (TNF-α).
The accumulation of lipids such as diacylglycerols (DAGs) and SFAs within macrophages, as a result of macrophage scavenging activity, has been implicated in developing a pro-inflammatory M1 macrophage phenotype. This lipid buildup can exert toxic effects on the endoplasmic reticulum, further exacerbating inflammatory responses in metabolic conditions [74, 75].
Omega-3 and omega-6 PUFAs are promising therapeutic targets for metabolic diseases and cardiovascular disease (CVD) due to their potent anti-inflammatory properties [76]. As essential fatty acids, PUFAs must be obtained through diet, and a high intake of PUFAs has been linked to a reduced risk of CVD, highlighting their regulatory role in inflammation [77]. The dysregulation of lipid metabolism in tumors supports cell proliferation and survival, as well as influences inflammatory pathways that contribute to carcinogenesis. Lipid metabolic signaling, through the crosstalk with pathways such as NF-κB and STAT3, can enhance the expression of pro-inflammatory cytokines like IL-6 and TNF-α, which in turn promote tumor progression and immune evasion [78, 79]. In PC, for example, altered lipid metabolism leads to the accumulation of lipid droplets, which can further activate inflammatory signaling pathways that create a pro-tumorigenic microenvironment [80].
Research on THP-1 macrophages has demonstrated that PUFAs, including linoleic acid, alpha-linolenic acid, and docosahexaenoic acid (DHA), can mitigate lipopolysaccharide (LPS)-induced inflammation [81]. Moreover, DHA decreases the production and secretion of pro-inflammatory cytokines IL-1β and TNF-α and enhances the secretion of the anti-inflammatory cytokine IL-10 through an autocrine mechanism. Furthermore, DHA activates key anti-inflammatory pathways involving peroxisome proliferator-activated receptor gamma ( [82] PPARγ ) [83] and AMP-activated protein kinase (AMPK), which in turn inhibit NF-κB, which is a major regulator of inflammation in macrophages [84].
Regarding PC, dysregulated lipid uptake through transporters such as CD36 and fatty acid-binding proteins (FABPs) facilitates the formation of lipid rafts [85, 86], which serve as platforms for the activation of oncogenic pathways such as PI3K/Akt and ERK [87, 88]. Additionally, enzymes involved in lipid metabolism, such as FASN and ACC, are often upregulated in early-stage pancreatic cancer, promoting cell survival and proliferation [89].
Metabolic reprogramming, inflammation, and immune suppression drive cancer progressionCancer cells require abundant energy to sustain their rapid growth, and lipids serve as a critical energy source that fuels cancer cell proliferation. Dysregulated lipid metabolism profoundly influences key metabolic pathways, such as increased de novo fatty acid synthesis and cholesterol biosynthesis, essential for supporting cancer cell growth [90]. These metabolic shifts allow the modification of lipids into molecules necessary for driving vital biochemical processes within the cell [91].
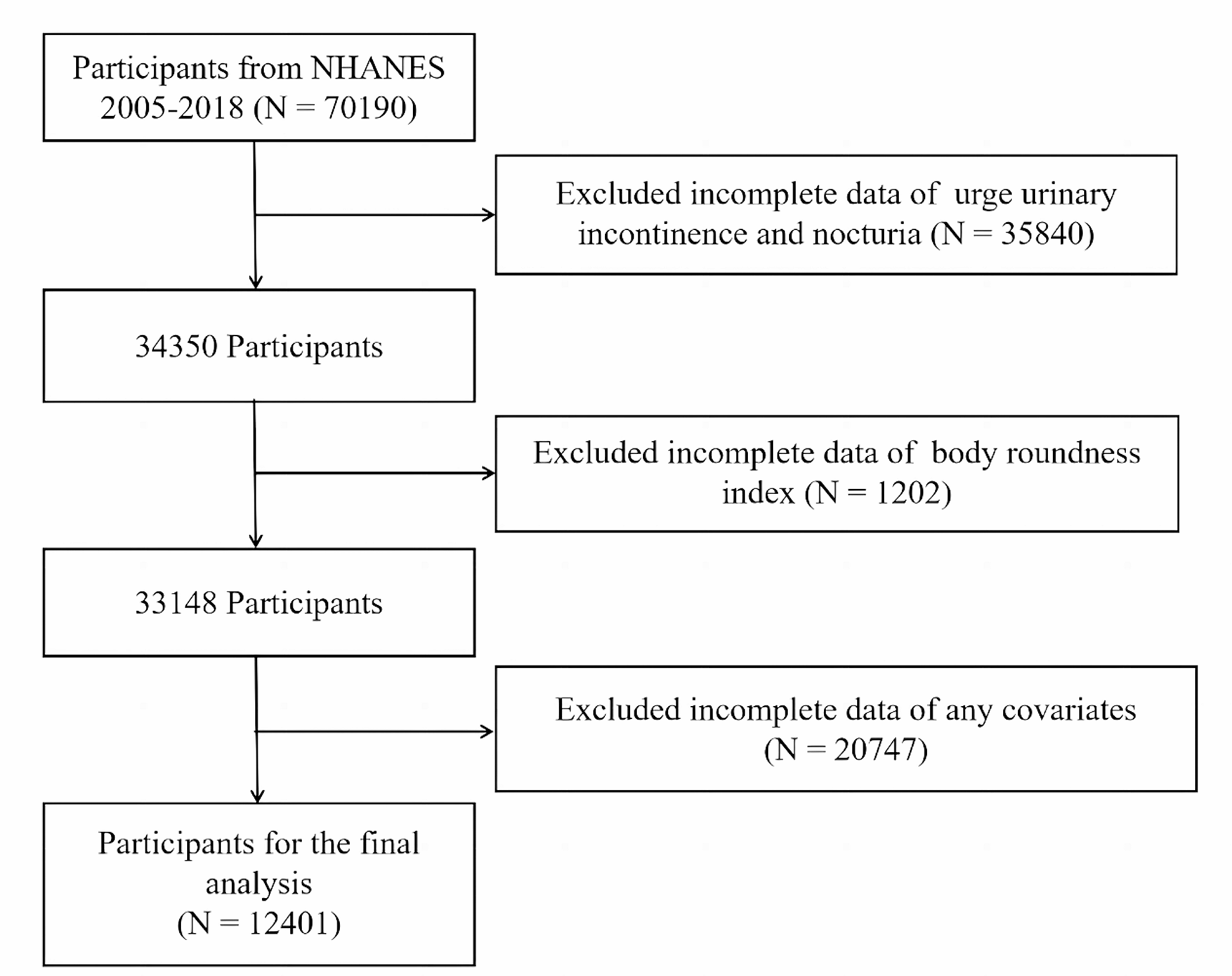
Altered lipid metabolism provides energy through FAO and glycolysis, supporting tumor progression by shaping immune responses, as shown in Fig. 1 [64]. This dysregulated lipid utilization contributes to chronic inflammation and immune suppression, creating a favorable environment for cancer cells to thrive and evade immune surveillance [92]. Therefore, lipid metabolism becomes intricately linked with tumor growth and cancer cells’ ability to bypass the immune system, reinforcing its critical role in cancer progression.
Lipids play a significant role in modulating the activity of myeloid-derived suppressor cells (MDSCs), critical for maintaining immune homeostasis, cell membrane integrity, and signaling. Research has shown that MDSCs isolated from cancer patients or tumor-bearing mice exhibit significantly higher lipid accumulation than controls [93], establishing a direct link between lipid buildup and immune suppression in cancer. This lipid buildup in MDSCs correlates with decreased T-cell activity, leading to immunosuppression [94,95,96]. It has been demonstrated that oxidized lipids serve as a key energy source supporting the immunosuppressive function of MDSCs within the tumor microenvironment. Specifically, MDSCs with excessive lipid content show a greater ability to suppress CD8 + T-cell activity compared to MDSCs with normal lipid levels [97]. A study by Cao et al. revealed that fatty acid transport protein 4 (FATP4) is overexpressed in tumor-derived MDSCs, suggesting a connection between lipid accumulation and increased fatty acid uptake [98]. This finding indicates that lipid metabolism in MDSCs is closely tied to their role in dampening immune responses, particularly in cancer settings.
Lipid accumulation is a major contributor to dendritic cell (DC) dysfunction in malignancies. Excess lipids in DCs impair their antigen-processing abilities, reduce the expression of costimulatory molecules like CD86, and increase the production of the tolerogenic cytokine IL-10, leading to immune tolerance as opposed to activation [99]. In ovarian tumors, the enzyme FASN, crucial for de novo lipogenesis, is upregulated. Consequently, these cancer cells produce more fatty acids, leading to higher fatty acid concentrations in the tumor microenvironment, which causes DCs to store excess fatty acids, thereby impairing their functionality [100].
Studies have also shown that the buildup of oxidized lipids, particularly triacylglycerol (TAG), further exacerbates DC dysfunction [101]. In ovarian cancer, lipid droplets accumulate within DCs, impairing their ability to mount effective anticancer responses. Another mechanism of DC impairment is the upregulation of carnitine palmitoyl transferase-1a (CPT1A), a key fatty acid transport protein, via the Wnt5a-β-catenin-PPARγ signaling pathway. This enhances FAO, promoting tumor resistance, as seen in melanoma [102].
Additionally, tumor-infiltrating DCs can trigger the endoplasmic reticulum (ER) stress response through the IRE1 protein, suppressing T-cell function and contributing to tumor growth [103]. These findings highlight how dysregulated lipid metabolism in immune cells, particularly in the tumor microenvironment, interferes with their ability to regulate carcinogenesis. The altered lipid metabolism in immune cells, influenced by their interaction with tumor cells, plays a pivotal role in undermining the anticancer immune response and facilitating tumor progression.
Lipids and their metabolites exert their anti-tumor effects also by regulating macrophage polarization. Lipogenesis is crucial for lipid accumulation and phagocytosis in M1 macrophages, whereas M2 macrophages depend on fatty acid β-oxidation as their primary energy source. Markers of lipolysis and fatty acid uptake, such as CD36, also contribute to cytokine production, leading to inflammation. Therefore, to strengthen the immune system by inhibiting lipid metabolism-related factors could represent a targeted approach against tumor cells.
In PC, metabolic reprogramming is tightly linked to inflammation and immune suppression, both of which accelerate tumor progression. Altered lipid metabolism drives chronic inflammation through the activation of pathways like NF-κB and STAT3 [104]. For instance, increased LDL-cholesterol levels have been shown to enhance pancreatic cancer cell proliferation, migration, and invasion by activating STAT3 phosphorylation, which promotes tumor survival and progression by regulating critical cancer hallmarks [105]. Additionally, research has demonstrated that overexpression of FASN in vitro induces resistance to genotoxic therapies by upregulating poly (ADP-ribose) polymerase (PARP)-1 expression and DNA repair mechanisms, through the NF-κB and specificity protein 1 (SP1) pathways in PC cells [106].
Enhancement of invasive and metastatic potential of cancer cellsLipids are essential for forming cell membranes and are crucial in enhancing cell motility and invasiveness, key factors in cancer metastasis [107]. Alterations in lipid composition can increase cancer cells’ ability to adhere to, invade surrounding tissues, and spread to distant organs. This lipid-driven enhancement of invasiveness is central to the metastatic process [108, 109]. Therefore, disruptions in lipid metabolism can destabilize these pathways, enhancing cancer cell invasion and migration, ultimately fostering cancer progression.
Two key products of lipid synthesis—FAs and cholesterol—play pivotal roles in metastatic cancers [110, 111]. For instance, chronic exposure to 27-hydroxycholesterol (27HC), an abundant cholesterol metabolite, selects for cells with increased lipid uptake and biosynthesis, enhancing their tumorigenic and metastatic capacity. This metabolic stress, driven by lipid accumulation, requires sustained GPX4 expression, a negative regulator of ferroptosis. Resistance to ferroptosis is a key feature of metastatic cells, and GPX4 knockdown reduces the tumorigenic and metastatic potential of 27HC-resistant cells [20]. In CRC, a cholesterol metabolic pathway specific to CRC liver metastasis has been identified, involving the activation of the SREBP2-dependent cholesterol biosynthesis pathway. This pathway is essential for the colonization and growth of metastatic CRC cells in the liver [112]. Anoctamin 1 (ANO1) has been identified as a key factor in driving metastasis in various metastatic cancer cell lines. ANO1 promotes cholesterol accumulation by inhibiting LXR signaling and reduces cholesterol hydroxylation by downregulating the expression of cholesterol hydroxylase CYP27A1. A novel small-molecule inhibitor of ANO1 has been shown to mitigate tumor burden at metastatic sites [113]. DHCR7 (7-dehydrocholesterol reductase), an enzyme that catalyzes the last step of cholesterol synthesis, has been reported to promote the invasion ability of cervical cancer cells and lymphangiogenesis in vitro and induced lymph node metastasis in vivo through cholesterol reprogramming-mediated activation of the KANK4/PI3K/AKT axis and VEGF-C secretion [114].
In hepatocarcinoma, the high expressed carnitine palmitoyltransferase 1 C (CPT1C) has been linked to increased cancer invasiveness and poor prognosis [115]. Reducing CPT1C levels downregulates FAO, impairing cancer cell growth, cell cycle progression, and metastatic potential [116, 117]. The role of FAO in tumorigenesis has also been highlighted in hepatocellular carcinoma, where increased FAO enhances cancer stemness. The overexpression of organic carnitine transporter 2 (OCTN2) is associated with poor prognosis in hepatocellular carcinoma. OCTN2 promotes tumor growth and invasion by enhancing FAO and mediating oxidative phosphorylation. Peroxisome proliferator-activated receptor-gamma coactivator (PGC-1α) upregulates OCTN2 expression, which in turn increases the expression of Yin Yang 1 (YY1), further driving cancer progression [118]. PRP19 promoted esophageal squamous cell carcinoma growth in vitro and in vivo by enhancing fatty acid synthesis through sterol regulatory element-binding protein 1 (SREBF1), a major transcription factor of lipid synthase [117]. These studies collectively demonstrate that dysregulation of FA metabolism, particularly through mechanisms like FAO, plays a critical role in promoting cancer metastasis and immune evasion.
Increased cholesterol metabolism can significantly enhance cancer progression. Inhibiting cholesterol metabolism in T cells has been shown to boost their anti-cancer cytotoxicity, whereas suppressing cholesterol metabolism in tumor cells can hinder their metastatic potential [119]. Targets of the liver X receptor (LXR), such as ABCA1 and ABCG1, are associated with increased cancer progression. For instance, in clear cell renal cell cancer, celastrol increases LXR, leading to increased expression of ABCA1, which affects cholesterol homeostasis and contributes to cancer migration and invasion [120].
Similarly, in breast cancer, nucleobindin-2 (NUCB2)/Nesfatin-1 upregulation correlates positively with prognosis and enhances cholesterol metabolism. NUCB2/Nesfatin-1 stimulates the mechanistic target of rapamycin complex 1 (mTORC1), promoting tumor cell invasion and metastasis [121]. Additionally, cholesterol metabolism impacts immune cell activity; X-box-binding protein 1 (XBP1), an oncogenic factor, increases cholesterol production and enhances the immunosuppressive function of myeloid cells [122].
In PC, lipids can also be tumor-stroma communication mediators for cancer and stroma cells. Mice with oncogenic KRAS and p53 mutations (KPC mice) fed a high-fat diet exhibit larger primary pancreatic tumors and increased metastasis compared to those on a standard diet. Fatty acids, likely derived from adipose tissue, are taken up by pancreatic cancer cells, promoting lipid droplet (LD) formation and enhancing tumor cell migration [123]. Additionally, a recent study demonstrated that berberine, an isoquinoline alkaloid with various pharmacological properties, significantly reduces acetyl-CoA carboxylase (ACLY) expression in the cytoplasm, disrupting lipid metabolism, thereby inhibiting pancreatic cancer cell proliferation and migration [124].
Overall, the interplay between cholesterol metabolism and cancer progression highlights the potential of targeting lipid metabolism as a therapeutic approach.
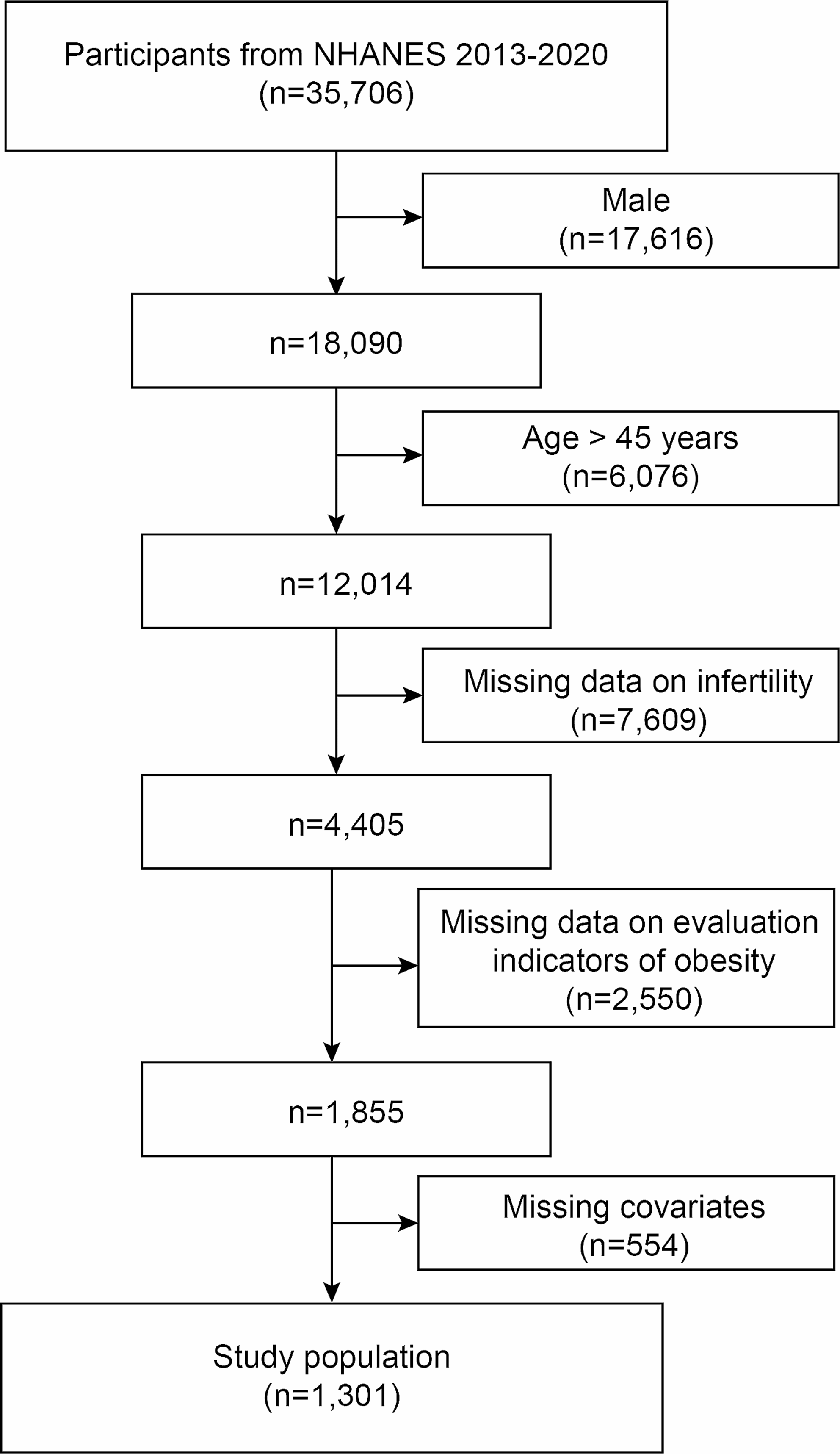
Lipogenic phenotype drives altered drug transport and enhances drug resistanceDysregulated lipid transporters significantly impact drug uptake and efflux, reducing drug efficacy. For example, the overexpression of ABC transporters is known to mitigate the effectiveness of therapeutic agents [125]. Lipids also modulate drug responses through various signaling pathways; sphingolipids, for instance, regulate apoptosis and drug sensitivity [126]. Additionally, lipid rafts—specialized membrane microdomains—affect the localization of drug receptors [127]. The altered composition of lipid rafts can contribute to drug resistance by changing receptor distribution and function. Figure 2 shows that elevated levels of cellular FAs and cholesterol drive tumor growth and reduce the effectiveness of therapeutic drugs. In tumor-infiltrating Treg cells, increased lipid synthesis, including FAs and cholesterol, is a key metabolic change, driven by the activation of SREBPs and their target genes [128]. Treg cells also promote FA and cholesterol metabolism through the AKT-mTORC1 signaling pathway, which supports cell proliferation and the expression of immune-suppressive molecules like CTLA-4 and ICOS [129]. Therefore, targeting lipid metabolism pathways in Treg cells, such as CD36 and SREBP, could enhance cancer immunotherapy efficacy and warrants further investigation [128, 130]. Natriuretic peptide receptor A (NPRA) was upregulated in gastric cancer cells cocultured with mesenchymal stem cells (MSCs), and the knockdown of NPRA reversed MSC-induced stemness and chemoresistance. NPRA facilitated stemness and chemoresistance through FAO by protecting Mfn2 from degradation and promoting its mitochondrial localization, and inhibition of FAO with etomoxir (ETX) reduced MSC-induced chemoresistance in vivo [131]. NSD2 promotes FAO by methylating AROS (active regulator of SIRT1) at lysine 27, facilitating the physical interaction between AROS and SIRT1, ultimately affecting the effectiveness of tumor radiotherapy [132].
In healthy tissues, lipid metabolism operates within a balanced framework, but tumor cells often exhibit dysregulated lipid metabolism to develop drug resistance, marking the lipogenic phenotype as a hallmark of malignancy. This phenotype is characterized by hyperactivation of FA synthesis pathways, promoting lipid accumulation that supports cancer progression [133].
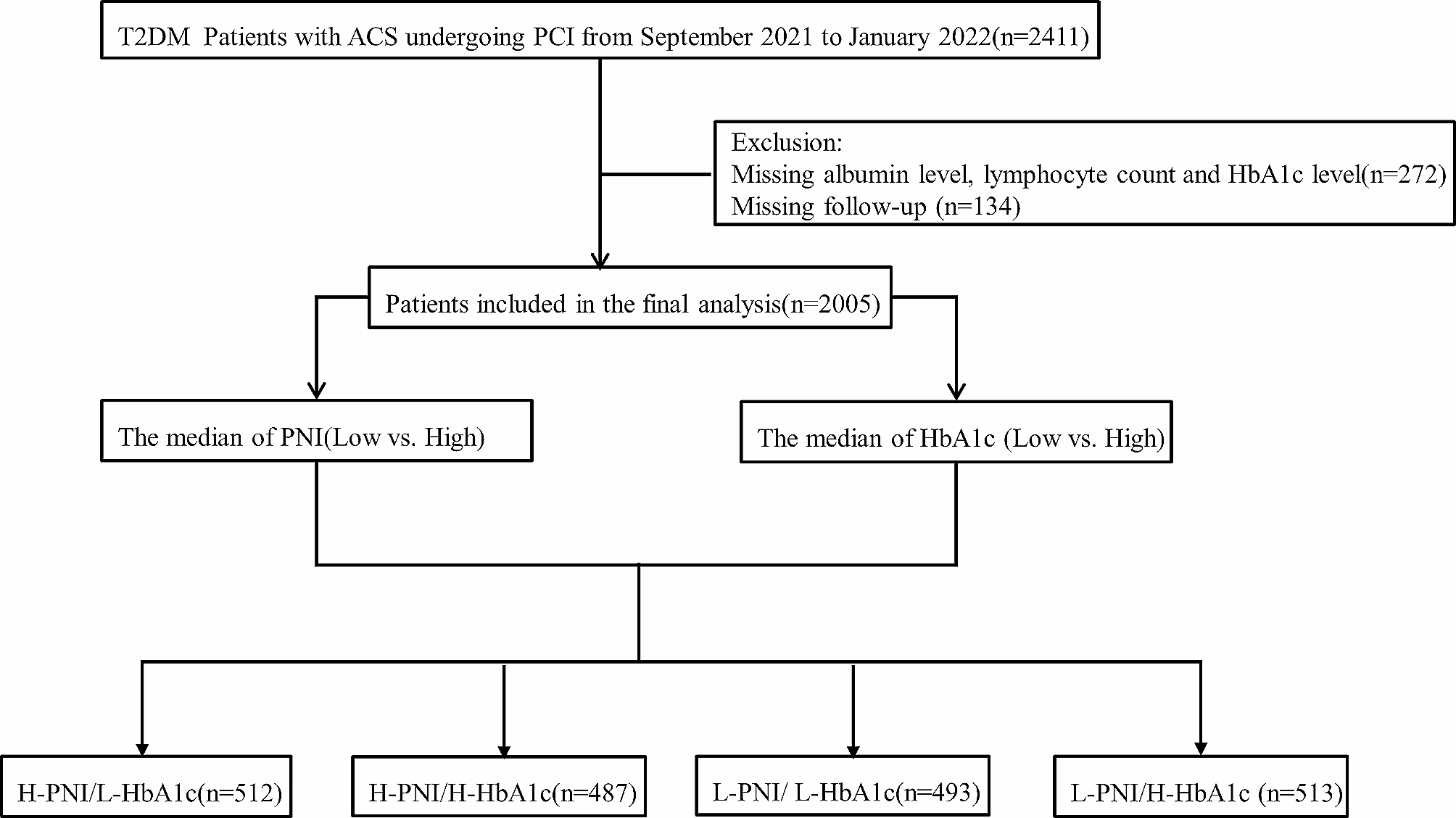
Increased lipid accumulation is observed in various cancers, including breast, colorectal, and ovarian, and correlates with poor prognosis. Elevated free FA levels in tumor cells enhance malignancy, with increased FA uptake activating the HIF1/MMP14 axis, driving invasion and metastasis [134,135,136]. Figure 3 illustrates lipid metabolism dysregulation in cancer.
Researchers have increasingly focused on the role of lipid metabolism in regulating PC progression, particularly regarding chemoresistance [137]. PC is characterized by rapid proliferation and metastasis, which often leads to resistance to therapy, especially in advanced stages [138]. Gemcitabine is the primary drug used to suppress PC, and understanding the molecular mechanisms behind gemcitabine resistance has been a key area of study [139]. Tadros [26] found that FASN expression increased with disease progression in a genetically engineered mouse model and was associated with poor survival and reduced gemcitabine responsiveness in human pancreatic cancer patients. FASN inhibitors, when combined with gemcitabine in both cell cultures and orthotopic models, reduced cancer stemness by inducing ER stress and apoptosis. Another study highlighted that TGFB2, stabilized post-transcriptionally by METTL14-mediated m6A modification, promotes lipid accumulation, with triglyceride buildup contributing to gemcitabine resistance as demonstrated by lipidomic profiling [140]. These findings collectively suggest that targeting lipid metabolism, particularly through inhibition of FASN and regulation of lipid accumulation, can enhance the effectiveness of gemcitabine in treating pancreatic cancer.
Lipid metabolism dysregulation has been recognized as a crucial factor in drug resistance development in PC [141]. Changes in lipid metabolism, alongside associated molecular interactions, can affect how PC cells respond to therapy. Besides, targeting lipid metabolism may also influence glucose metabolism in PC cells, potentially altering their response to chemotherapy.
Dysregulated lipid metabolism plays a vital role in key processes such as cancer initiation, progression, metastasis, and drug resistance, reflecting its complex connection with cancer biology and its potential as a critical therapeutic target. However, most studies on the association between lipid metabolism and cancer are conducted in cell or animal models, leaving significant uncertainties about their applicability in clinical settings. For instance, while inhibiting specific lipid metabolic enzymes shows efficacy in in vitro studies, the complexity of the human body might alter enzyme functions and influence therapeutic outcomes. Bridging this gap between experimental findings and clinical applications remains a significant challenge.
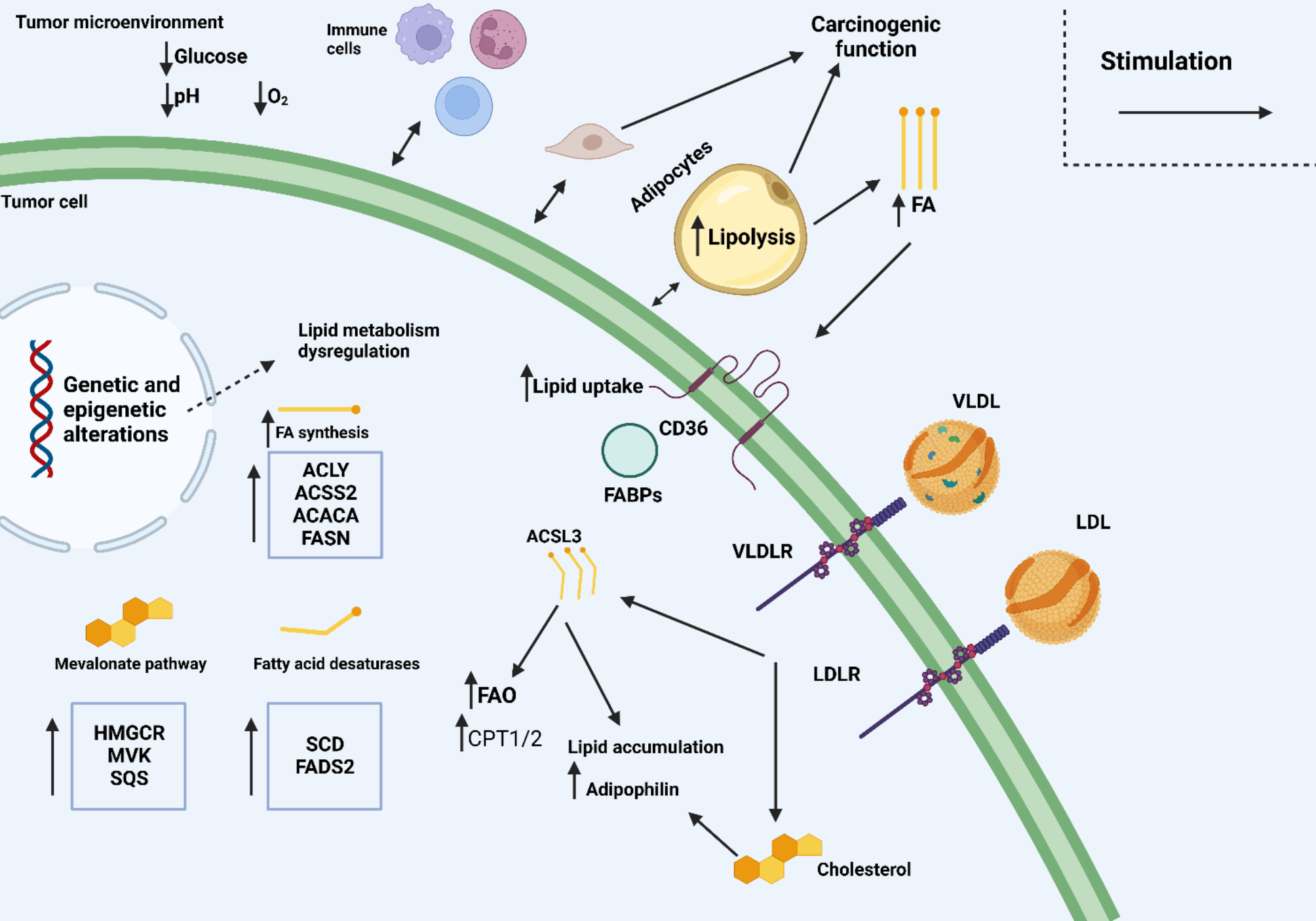
Fig. 1
Reprogramming of lipid metabolism in cancer
Tumor cells frequently reprogram their lipid metabolism to enhance their survival and growth under adverse conditions. This metabolic adaptation arises from genetic and epigenetic changes and interactions with the tumor microenvironment (TME). Within the TME, tumor cells influence and are influenced by various components, resulting in alterations in multiple metabolic pathways. One significant example is the induction of lipolysis in neighboring adipocytes, which release fatty acids (FAs) that are subsequently absorbed by cancer cells. This process supports lipid accumulation and FAO in tumor cells. Tumor cells can also assimilate lipoproteins from external sources, further contributing to their lipid metabolism. Additionally, while immune cells in the TME can possess anti-tumoral properties, tumors often subvert their functions, converting them into suppressive or pro-tumorigenic entities. Tumor cells produce lipid mediators that impact tumor progression by influencing angiogenesis and immune responses. Lipid metabolism reprogramming varies across different tumor types, reflecting the complexity and diversity of these mechanisms. Key molecules and pathways involved in this process include ACACA (acetyl-CoA carboxylase 1), ACLY (ATP-citrate lyase), ACSL3 (acyl-CoA synthetase long-chain family member 3) ACSS2 (acetyl-CoA synthetase 2), CAFs (cancer-associated fibroblasts), CPT1/2 (carnitine palmitoyltransferase 1 and 2), FABPs (fatty-acid-binding proteins), FADS2 (fatty acid desaturase 2), FASN, HMGCR (HMG-CoA reductase), LDL/LDLR (low-density lipoprotein/receptor), LPA (lysophosphatidic acid), LPL (lipoprotein lipase), MVK (mevalonate kinase), PGE2 (prostaglandin E2), SCD (stearoyl-CoA desaturase), SQS (squalene synthase), VLDL/VLDLR (very-low-density lipoprotein/receptor). This comprehensive overview emphasizes the critical role of lipid metabolism in cancer progression and highlights the diversity of metabolic adaptations employed by tumor cells [142].
Fig. 2
Role of lipids and fatty acid metabolism in cancer development. Elevated levels of cellular FAs and cholesterol are associated with enhanced cancer progression. Increased FA and cholesterol metabolism drive tumor growth and contribute to reduced efficacy of therapeutic drugs [54]. This metabolic dysregulation also exacerbates the immunosuppressive functions of myeloid cells, potentially impairing T-cell function [143, 144]. The impact of elevated lipid metabolism on immune responses and drug efficacy underscores the critical need to target lipid metabolic pathways to combat chemoresistance. Cancer treatments aim to overcome resistance and improve therapeutic outcomes by inhibiting FA and cholesterol metabolism
Fig. 3
Comments (0)