
Remember me




Vascular Thiol Isomerases (VTIs) are multifaceted enzymes possessing oxidoreductase, isomerase, and chaperone functions, essential for a variety of cellular processes [1]. Characterized by the presence of endoplasmic reticulum (ER) retrieval motifs (exemplified by the KDEL sequence in PDI), these enzymes are predominantly localized within the ER, where they facilitate the formation of native disulfide bonds in newly synthesized proteins and contribute to quality control of client proteins [2]. In response to vascular injury, activated platelets and endothelial cells release several VTIs into the extracellular milieu [3, 4]. These released VTIs exert a significant influence on thrombus formation by modulating the activities of vascular cell receptors [5,6,7], adhesive proteins [8,9,10], and coagulation factors [11,12,13]. In this review, we summarize the structures and functions of VTIs, elucidate their intricate role in thrombus formation, and highlight progress in the development of anti-thrombotic compounds specifically targeting VTIs.
Structures and functions of VTIsPDI represents the first VTIs recognized for its pivotal role in vascular biology [14, 15]. Subsequently, ERp5, ERp46, ERp57, ERp72, TMX1, and TMX4 were found to serve important roles in the regulation of thrombosis [16, 17]. The unique structures and redox potentials of these VTIs contribute to their different functions in thrombosis.
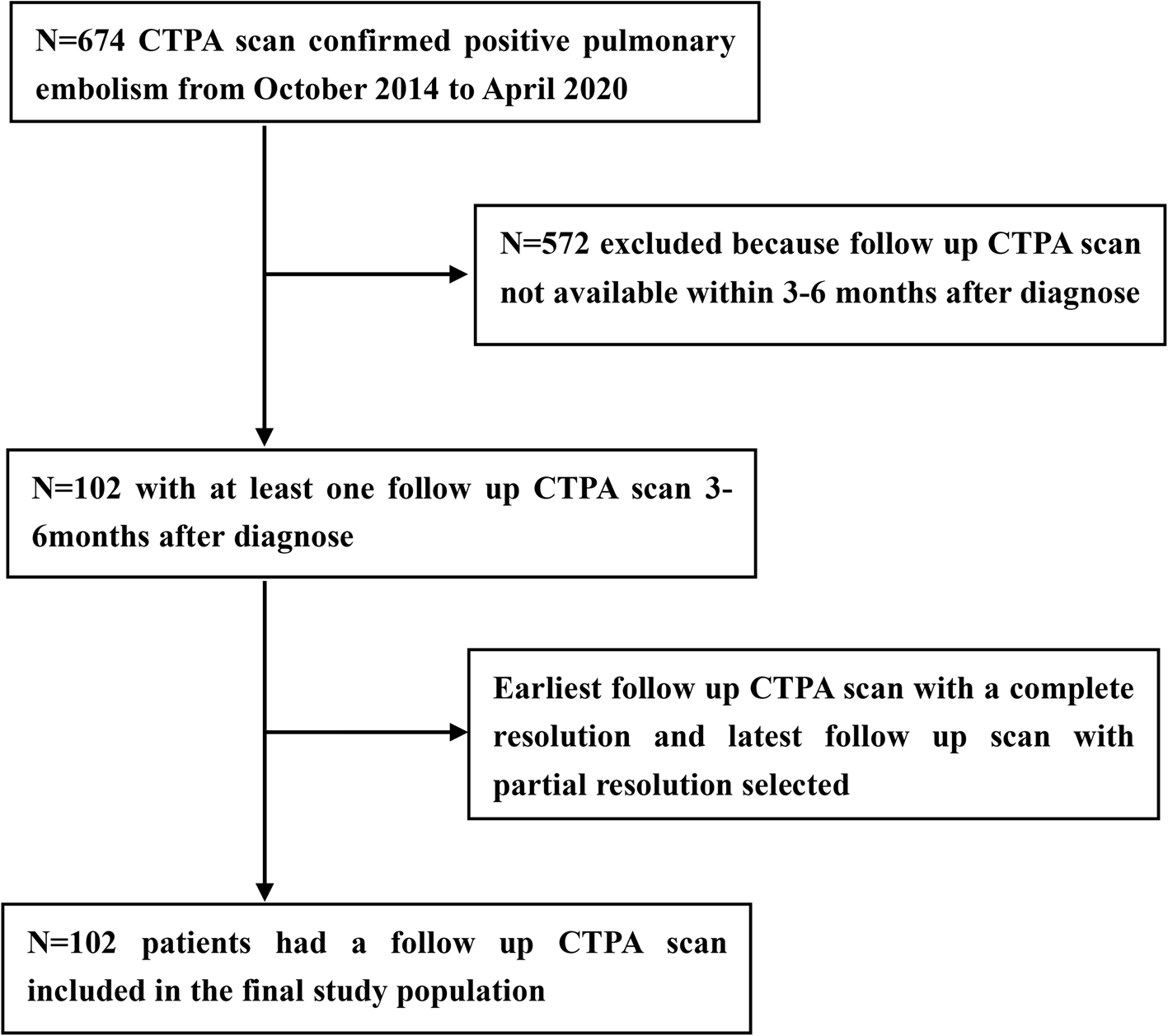
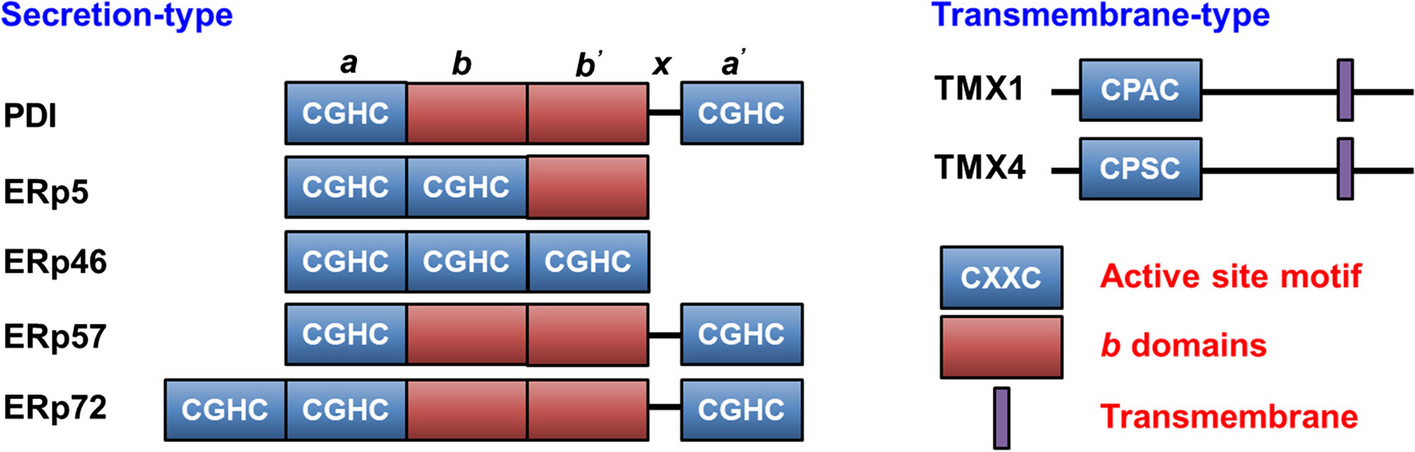
Structurally, these VTIs are characterized by the inclusion of at least one thioredoxin-like domain, which is comprised of a four-stranded antiparallel β-sheet encircled by three α-helices. These thioredoxin-like domains are subclassified into catalytic domains (a0, a, and a’ domains) containing the CXXC motif (where X denotes any amino acid), and non-catalytic domains (b and b’ domains) [18]. The CXXC motif within the catalytic domain is indispensable for the functional activity of VTIs (Fig. 1). Specifically, the two cysteine residues within this motif endow the enzyme with diverse redox capabilities [19]. The N-terminal cysteine of the CXXC forms a mixed heterodimer with the substrate, while the C-terminal cysteine facilitates substrate release. The amino acid residues positioned between the cysteine residues determine the redox potential and functional specificity of the enzyme [20]. Conversely, the b and b’ domains serve as spacers and are primarily involved in substrate recruitment.
Fig. 1
B Representative vascular thiol isomerases (VTIs) and their domain organization, with a-type domains in darkblue, b-type domains in darkred, and transmembrane domains in purple. A 19-amino acid linker (X-linker) connects the b’ and a’ domains and affects substrate binding
In addition to their intricate structural features, VTIs exhibit a diverse range of functions that are crucial for cellular homeostasis and disease processes. Within the endoplasmic reticulum (ER), these enzymes are indispensable for the proper folding and assembly of nascent proteins by facilitating the correct formation of disulfide bonds. However, their functions extend beyond the ER, as thiol isomerases have been implicated in various extracellular and cytosolic processes. For instance, thiol isomerases such as PDI have been shown to possess reductase and oxidase activities, which are vital for regulating the cellular redox status and protecting against oxidative stress [21]. Furthermore, recent studies have highlighted the role of thiol isomerases in pathological conditions such as thrombosis, where they interact with platelet receptors and coagulation factors to modulate blood clot formation [22, 23]. These findings underscore the complex and multifaceted nature of VTIs and their importance in maintaining cellular health and preventing disease. By gaining a deeper understanding of their functions and regulatory mechanisms, researchers can potentially identify novel therapeutic targets for the treatment of disorders associated with VTIs dysregulation.
PDIPDI contains four thioredoxin-like domains, namely a, b, b’, and a’. The a and a’ domains are the catalytic domains, and their characteristic CGHC motifs (WCGHCK) are responsible for forming, breaking, and rearranging disulfide bonds [24, 25]. The catalytic domains are separated by b and b’ domains, with the b’ domain serving a dominant role in substrate binding [26]. A 19-amino acid linker (X-linker) connects the b’ and a’ domains and affects substrate binding [27]. The C-terminal of PDI contains a large number of negatively charged amino acids, which play an important role in the chaperone function of PDI [28]. Crystal structural studies show that PDI is arranged in a U-shape. The catalytic domains a and a’ are located at the two ends of the U-shaped structure. The non-catalytic domains b and b’ are distributed in the bottom area, with a hydrophobic pocket in the b’ domain for substrate binding [29]. PDI not only functions as a single individual protein, but is also the β subunit of prolyl 4-hydroxylase and microsomal triglyceride transfer protein (MTP) [30]. The structure of MTP reveals the molecular mechanisms of PDI interaction with large protein substrates [31]. The b’ domain of PDI provides a primary binding site for MTPα, together with both catalytic domains, mainly through hydrophobic interactions, further supporting the premise that the b’ domain of PDI is the principal substrate-binding site, but all domains together contribute to the binding of substrate [18].
ERp57Similar to PDI, ERp57 consists of four thioredoxin-like domains and forms a similar U-shaped structure [32, 33]. Unlike the b’ domain of PDI, which has a hydrophobic pocket to bind substrate, the corresponding pocket of the ERp57 b’ domain is positively charged. This pocket is critical for ERp57 to interact with negatively charged substrates, such as the arm-like P-domain of lectin chaperones calnexin (CNX)/calreticulin (CRT) [34] to facilitate the correct folding and the quality control of neo-synthesized glycoproteins [35]. The b domain enhances ERp57’s binding to the substrate [36], and the basic C-terminus of ERp57 is also responsible for the specific interactions with negatively charged partners. ERp57 engages in the peptide-loading complex (PLC) formation by interaction with CRT and tapasin [37]. The b’ domain of ERp57 interacts with CRT to form a flexible belt around tapasin and major histocompatibility complex class I (MHC-I), which play key roles not only in catalyzing the oxidative folding of glycoproteins, but also in stabilizing the PLC to control peptide loading [33]. ERp57 interacts with tapasin, particularly with a and a’ domains via hydrophobic interactions, analogous to how the a and a’ domains of PDI interact with MTP [31]. However, in this case, a mixed disulfide bond forms between ERp57 and tapasin.
ERp72ERp72 is the only VTI with five thioredoxin-like domains, which is due to an additional N-terminal catalytic domain a0 to give an a0-a-b-b’-a’ architecture. Small-angle X-ray scattering (SAXS) studies show that the overall structure of ERp72 is crescent-shaped [38]. Site-directed mutagenesis in the N-terminal Cys of these catalytic domains indicates that the a0 domain is primarily involved in catalysis, the a domain has intermediate roles in catalysis and binding, and the a’ domain functions primarily to bind substrates [39]. The X-ray crystal structure of ERp72 bb’ reveals that these two domains form a rigid pair due to the lack of a flexible interdomain linker. The b’ domain of ERp72 has a negatively charged patch. The residues Arg398 and Glu459 form a salt bridge to occlude this potential substrate-binding cavity, making it neither binding to hydrophobic substrates like PDI nor negatively charged partners like ERp57 [38]. Alternatively, the X-ray crystal structure of the ERp72 a0a fragment reveals both two domains contain a small hydrophobic patch adjacent to the catalytic sites [40]. A structural model of full-length ERp72, using the a0a domains together with bb’ and a’ domains, shows that all three catalytic sites can be positioned to face each other, and the hydrophobic patches adjacent to the catalytic sites are available for protein substrate binding [40]. Such hydrophobic patches are also found in other VTIs, which cooperatively bind to substrate [41, 42]. Similar to PDI, ERp72 also contains a large number of negatively charged amino acids at its terminus, but these residues are located at the N-terminus of ERp72, which are considered to be the binding site of Ca2+, and mediate ERp72’s interaction with chaperones such as BiP, GRP94, and cyclophilin B [43]. Also, one well-defined substrate for ERp72, NADPH oxidase 1 (NOX1), specifically interacts with the first 105 residues of ERp72, which contains this putative Ca2+ binding sequence and part of the a0 domain [44].
ERp46ERp46 belongs to the protein disulfide isomerase (PDI) family, characterized by the presence of three surface-exposed CGHC motifs located within the a0, a, and a' domains, which are delineated by flexible linker loops [45]. ERp46 has a radically different molecular architecture compared to other PDIs. This architectural feature distinguishes ERp46 from the more compact, U-shaped conformations of PDI, ERp57, and ERp72, where the active sites are juxtaposed, facing each other across the substrate-binding cleft. Notably, the active sites of ERp46 function independently, contrasting with the cooperative activity displayed by the active sites of PDI. ERp46 exhibits a rapid and promiscuous capacity to introduce disulfide bonds into unfolded substrates, followed by a subsequent, slower, and more precise disulfide bond formation mediated by PDI [46]. Expression of ERp46 on the platelet surface increases with thrombin stimulation. Functionally, ERp46 serves a protective role against apoptosis and is implicated in the production of immunoglobulins and insulin, as well as in the growth of prostate cancer cells [47]. Despite the observations by Holbrook et al. that ERp46 is expressed in human megakaryocytes and human umbilical vein endothelial cells [48] and by Zhou et al. that ERp46 functions in platelet activation and thrombus formation [49], the precise role of ERp46 in the context of thrombosis remains elusive.
ERp5ERp5 contains two catalytic domains (a0 and a’), and one noncatalytic b domain, organized in a unique a0-a’-b arrangement. Although the overall structure of ERp5 is still not determined, ERp5 may form an opened and extended conformation like ERp46, which allows easy access for clients, facilitating disulfide bond introduction into clients during the early stages of oxidative protein folding [50]. The crystal structure of ERp5 a0 domain in a complex with the Prx4 C-terminal peptide [51] showed the peptide bound a hydrophobic groove of ERp5 a0 adjacent to the redox-active cysteine, which also exists in other VTI family proteins [41, 42].
TMX1TMX1 (thioredoxin-related transmembrane protein 1, alternative name TXNDC1) is the first identified transmembrane member of the VTI family [52], and the best-known member of the TMX family [53]. TMX1 is a single-pass type I membrane protein of 280 residues with a large luminal N-terminal region harboring a TRX-like domain and a short cytosolic tail. TMX1 displays a non-canonical CPAC active site in its type-a TRX-like domain [53]. The cytosolic tail of TMX1 also contains both palmitoylation and phosphorylation sites [54]. TMX1 does not contain the b or b’ domain and typical ER-retention sequence as other VTIs. In contrast, the di-arginine (RQR) motif in the cytosolic tail may promote its retention within the ER. TMX1 is critical for maintaining platelets in a quiescent state and counterbalancing the effect of ERp46 to prevent platelet overactivation [55]. TMX1 has been also found to interact with vitamin K epoxide reductase (VKOR), an enzyme involved in the process of blood coagulation working with membrane-tethered TRX-like proteins, which serve as redox partners [56].
TMX4TMX4 (thioredoxin-related transmembrane protein 4, alternative name TXNDC13) is a single-pass type I glycoprotein of 349 amino acids that was identified in 2010 during a database search for TRX-like domains containing proteins [57]. Phylogenetic analysis showed that TMX4 represents the paralogue of TMX1, with whom it shares high similarity within the N-terminal luminal regions despite the presence of an N-glycosylation site and a di-arginine RQR retention motif within the C-terminal domain [58]. Consistently with the lack of an ERSE motif within its promoter region, TMX4 is not up-regulated during ER stresses. TMX4 has one luminal type-a TRX-like domain, which contains a non-canonical CPSC active site. Supporting an involvement in protein folding, TMX4 interacts with CNX and with ERp57 [59]. In this functional complex, TMX4 could enzymatically modify clients directly promoting their oxidative maturation, or it could indirectly contribute to protein folding by reducing the ERp57 catalytic site thus promoting its function as a glycoprotein-specific oxidase.
VTIs and substrates in thrombosisVTIs are localized to the endoplasmic reticulum of megakaryocytes [60, 61] and the dense tubular system in platelets [61, 62]. In endothelial cells, VTIs are localized to vesicles distinct from Weibel-Palade bodies [63, 64]. Activating platelets or endothelial cells releases VTIs onto cell surfaces and into circulation [22, 48]. Circulating VTIs are thought to regulate the activity of vascular cell receptors, adhesive proteins, and clotting factors on activated platelets and endothelial cells by affecting their allosteric disulfide bonds and regulating vascular events, including blood coagulation, platelet activation, and thrombosis [14].
Studies using targeted knockout mice, specific antibodies, and inactive recombinant proteins have provided hints regarding the role of VTIs in thrombosis. Thrombus formation is impaired in genetically engineered mice with either deletions or function-blocking mutations in PDI, ERp46, ERp57, and ERp72 [49, 65,66,67]. Inhibitory antibodies to PDI, ERp46, ERp57, ERp72, or ERp5 substantially reduced thrombus accumulation and fibrin generation in mice [49, 68,69,70,71,72]. Several inactive recombinant VTIs with mutations at the cysteines in the CXXC motifs were shown to interfere with the formation of thrombus [65, 66, 70, 73]. In contrast, TMX1 was shown to have a negative effect on thrombosis [74]. In TMX1-knockout mice, platelet aggregation, ATP release, activation ofαIIbβ3, and P-selectin expression increased. Inhibitory anti-TMX1 antibody increased platelet aggregation and ATP release induced by agonists of the GPVI and thrombin receptors. The recombinant extracellular domain of wild-type TMX1 (rTMX1) inhibits platelet aggregation and ATP release, while the inactive rTMX1 mutant potentiated these processes [75].
Extensive experiments were carried out to identify the substrates of VTIs. VTIs were shown to regulate the activity of integrins (αIIbβ3) [76], glycoprotein Ibα (GPIbα) of the GPIb-IX-V complex [77], thrombospondin-1 (TSP-1) [8], vitronectin, coagulation factors, and other proteins which are secreted from platelet α-granules and participate in the formation of thrombus [78]. VTIs represent a critical mechanism regulating the functions of hemostatic proteins. Histidine-rich glycoprotein (HRG) has been reported as a substrate of PDI and the procoagulant and anticoagulant activities of HRG were enhanced by the neutralization of endothelial heparan sulfate (HS) and inhibition of factor XII (FXIIa) activity in a recent study [79]. Thus, the PDI-HRG pathway fine-tunes thrombosis by promoting its rapid initiation by neutralizing HS and preventing excessive propagation by inhibiting FXIIa.
Platelet surface receptorsIntegrin αIIbβ3 is the most abundant receptor on the platelet surface [76]. On resting platelets, inactive αIIbβ3 adopts a bent conformation burying the headpiece to prevent ligand interaction. Upon activation by agonists, αIIbβ3 undergoes a conformational change from a bent to an extended form [80], exposing its headpiece that recognizes RGD and KQAGDV peptides present on fibrinogen, von Willebrand Factor (vWF) and fibronectin, and crosslinking with platelets for their adhesion, aggregation, and clot formation at the site of vessel injury [81, 82]. The extracellular region of β3 has four epidermal growth factor (EGF)-like domains [83]. The disulfides in the EGF domains are generally important for maintaining the inactive state of β3. For instance, Cys560-Cys583 in EGF-4 was identified as an allosteric disulfide bond for αIIbβ3 [84].
A recurring theme of the VTIs, including ERp5, PDI, ERp57, ERp72, ERp46, TMX1 and TMX4, is their influence on the function of αIIbβ3 [23]. One clear mechanism is ERp5 in the regulation of the activity of αIIbβ3 [85]. ERp5 binds to β3 integrin with a KD of 21 μM independent of Mn2+. The cysteine residues in the ERp5 active sites are not required for binding to β3 integrin [70]. Interestingly, ERp5 can cleave the disulfide bond between Cys177 and Cys184 at the edge of the fibrinogen binding pocket on the βI domain of αIIbβ3 in a force-mediated manner, thereby regulating the release of fibrinogen from αIIbβ3 and weakening the adhesion of platelets [7]. The stretching of the disulfide bond by either ligand binding or mechanical force is necessary for ERp5 activity. It has been proposed that this chemical event may limit thrombus growth and thus prevent vessel occlusion. It seems counter-intuitive that ERp5 promotes the release of fibrinogen from platelets during clot formation, yet ERp5 induces thrombosis in animal models. A possible explanation is fibrinogen released from αIIbβ3 promotes the binding of other ligands such as vWF to form more stable adhesion under high shear force [7, 86].
Regulatory roles of other VTIs on the ligand-binding activity of α
Comments (0)