
Remember me




One hundred and nine (109) women were identified by our combined search strategy. Among them, 72 (66%) resulted to be heterozygous for DMD mutations. Based on the availability of clinical and/or cardiological data and DNA samples, we selected 47 DMD heterozygous women for further studies. Among them, 38 subjects were unrelated while 9 were distributed across 4 families. None of the individuals included were of black ethnicity.
DMD mutationsHeterozygous DMD gene mutations were identified in all females (Supplemental Table S1). As expected, single- or multi-exon out-of-frame deletions represented the majority of mutations (20/47; 43%), followed by nonsense mutations (11/47; 23%) and out-of-frame duplications (6/47; 13%). In the remaining patients, we identified small out-of-frame deletions (4/47; 8%), intronic mutations (5/47; 11%) predicting aberrant splicing with disruption of the open reading frame, and one large DMD gene rearrangement (1/47; 2%).
DMD carriers’ cohortOf the 47 females meeting our selection criteria, 27 (57%) were asymptomatic carriers (grade 0), while 20 (43%) exhibited symptoms of dystrophinopathy. According to phenotype classification among the symptomatic women, 7 showed a mild (grade 1), 6 an intermediate (grade 2), and 7 a severe phenotype (grade 3); none were DMD-like (grade 4). The global mean age at the last visit was 47.2 ± 14.4 years, 40.7 ± 10.4 years for the asymptomatic carriers, and 55.75 ± 14.4 years for the symptomatic participants (mild were 47.43 ± 15.27, intermediate were 57.80 ± 17.06, and severe DMD heterozygotes were 62.30 ± 6.52 years old). There was a significant correlation between age at the last visit and clinical severity (Spearman’s rank correlation rho = 0.63, p < 0.0001).
Clinical and genetic data are summarized in Supplemental Table S1. The age at last evaluation ranged from 14 to 80 years. Family history was positive in 38 (80%) cases. One patient with severe muscular weakness had died at the age of 65 years due to cardiac failure (#27).
Twenty (43%) heterozygous females exhibited muscle weakness (Supplemental Table S2). Age at symptom onset varied from 8 to 69 years (mean: 39.8 ± 16.7). In detail, mean age of onset was 32.9 ± 13.1 years in severely affected patients, 43.0 ± 21.0 in intermediate, and 44.0 ± 16.1 in mild patients. Proximal muscle weakness in the lower limbs (15/20; 75%) was the most common presenting symptom along with difficulty in running (4/20; 20%), jumping (1/20; 5%), or climbing stairs (3/20; 15%). Among first symptoms, myalgia was frequently reported (6/20; 30%), accompanied by other milder muscle symptoms such as subjective fatigue and/or muscle weakness. All patients but one (#3) were ambulant at last clinical evaluation and five (patients #5, #10, #11, #29 and #35) displayed severe impairment of ambulatory function, as they were only able to walk with support. Five of 7 severely affected women had lost the ability to rise from the floor. Asymmetry, defined as asymmetric muscle weakness upon physical examination, was observed in six patients (#5, #27, #29, #35, #36, #45). Of these individuals, four exhibited greater weakness on the right side and two on the left.
Gluteus, iliopsoas, quadriceps femoris, and deltoid were the most prominently affected muscles, as expected in dystrophinopathies. However, in our cohort, distal weakness was often observed, especially in severe (6/7) and intermediate (3/6) patients. Axial muscles were also frequently involved (12/20). Macroglossia was observed in four symptomatic patients and in one who was asymptomatic. Calf hypertrophy was a frequent clinical feature both in symptomatic (11/20; 55%) and asymptomatic women (6/27, 22%).
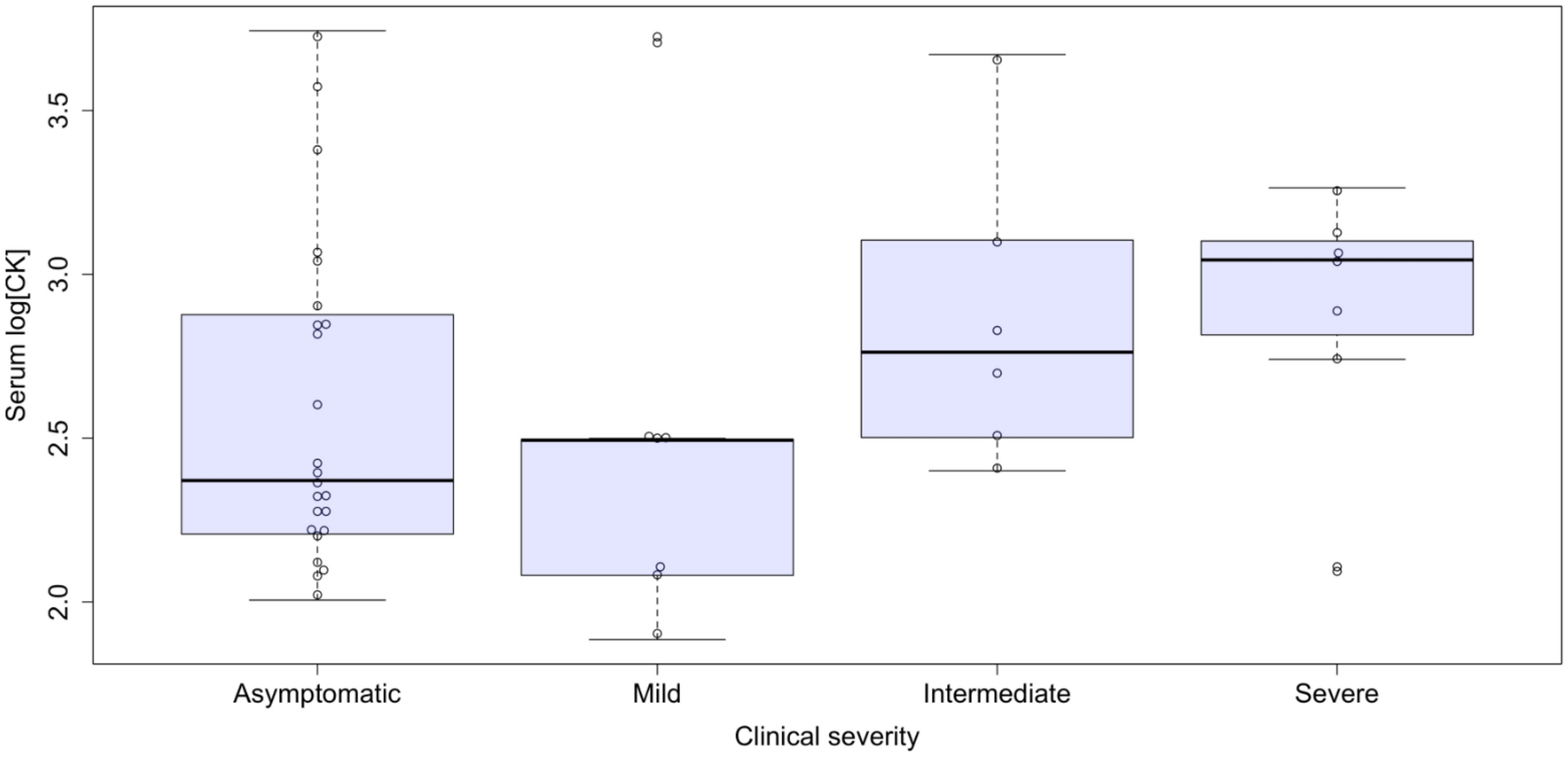
CK levels were available in 44 participants. High serum CK levels (defined as above 217 U/L) were detected in 13 (13/24; 54%) asymptomatic carriers and 16 (16/20; 80%) symptomatic patients (Supplemental Table S1), with mean values of 812 ± 1275 U/L and 1037 ± 1378 U/L, respectively (p = n.s.). An ordinal regression analysis for CK levels did not reveal a statistically significant association with clinical severity (Fig. 1).
Fig. 1
Serum CK levels (UI/L, log base 10) according to skeletal muscle phenotypes. Ordinal logistic regression: p = n.s
Cardiac functionCardiac data was available in 44/47 patients and is summarized in Supplemental Table S3. Cardiac involvement in terms of LV dilatation/dysfunction was found in 9 women: 3/7 (43%) of severely, 3/6 (50%) of intermediate, and 2/7 (29%) of mildly affected patients, and in 1/23 (4%) of the asymptomatic carriers. The mean age at detection of cardiac involvement was 56.2 ± 14.9 years (min 32, max 74 years). Thirteen patients (28%) received a cardiological therapy, including angiotensin-converting enzyme inhibitors (ACEi), beta blockers (BB), diuretics, calcium channel blockers (CCB), angiotensin receptor blockers (ARB) and anticoagulants, most often as combination therapy that included two or more medications. Nine out of these had a cardiomyopathy, while subjects #2, #7, #35 and #36 were only treated for high blood pressure.
By means of echocardiographic analysis, a LV dilatation was detected in five (5/44, 11%) patients (mean total cohort EDVi 54.5 ± 15.5 ml/m2, range 33–122 ml/m2), while six subjects (6/44, 13.6%) exhibited a LV-systolic dysfunction (mean total cohort LV-EF 58.4 ± 8.7%, range 32-78%; Supplemental Figure S1 and S2; Supplemental Table S3). There was a significant negative correlation between age and LV-EF (Spearman’s rank test rho = − 0.35, p = 0.02), while no correlation was found with EDVi (rho = − 0.02, p = 0.9). In 9/40 (22.5%) cases wall motion alterations (WMA) were detected, mainly in basal segments of posterior and inferior walls. Concerning the right ventricle (RV), a dilation was observed in 2 patients (2/44, 5%; mean end-diastolic area [RV-EDAi] of 8.88 ± 2.39 cmq/m2, range 6–19 cmq/m2), while a dysfunction in one case (1/44, 2%; mean fractional area change [FAC] of 47.6 ± 12.2%). None presented with RV-WMA. Fifteen patients (5 symptomatic and 10 asymptomatic) underwent myocardial strain analysis, and abnormal values were detected in two symptomatic (2/5; 40%) and in two asymptomatic women (2/10; 20%). Given the known association between high blood pressure and impaired global myocardial systolic strain, we ruled out hypertension in three of the four patients with strain abnormalities.
ECG was available in 42 patients (89%). All had sinus rhythm except one with persistent atrial fibrillation. Two patients had intraventricular conduction delay (incomplete right bundle branch block and anterior fascicular block, respectively). No patients had ventricular repolarization abnormalities. On 24-hour Holter ECG monitoring, three patients showed supraventricular ectopic beats, and another three showed premature ventricular beats. Moreover, two patients required implantable cardioverter defibrillator (ICD) implantation. More specifically, patient #21 was implanted in secondary prevention due to an episode of sustained ventricular tachycardia (SVT), whereas patient #27 was implanted in primary prevention at the age of 63, two years before her death for heart failure due to dilated cardiomyopathy with a severe reduction of LV systolic function.
Symptoms of heart failure were rare, only reported by these two patients with severe cardiomyopathy (#21, #27).
Dystrophin protein studiesIn our cohort, six asymptomatic carriers and nine symptomatic patients underwent a diagnostic muscle biopsy; in asymptomatic carriers, the indication was elevated CK. All biopsies were analyzed by dystrophin immunoblotting assay, and 12 also by immunofluorescence analysis.
Dystrophin immunofluorescenceAll biopsied patients showed a mosaic of dystrophin positive and negative fibers (Fig. 2). All muscle fibers in muscle cryosections stained with an anti-dystrophin antibody were counted on photographic enlargement and used to calculate the percentage of dystrophin positive and negative fibers (Supplemental Table S4). The mean percentage of dystrophin negative fibers was 32 ± 41.3 in symptomatic patients, and 11.5 ± 2.9 in asymptomatic carriers.
Fig. 2
Immunofluorescence analysis shows a mosaic pattern of dystrophin positive and negative fibers in DMD heterozygous females. Cryosections were stained with anti-dystrophin antibody NCL-DYS2 (panel A and C) and, as an example, some dystrophin negative fibers are indicated by an asterisk. A Immunolabeling of a 4-year-old girl (patient #43) and C of a 2-year-old girl (patient #9) showing dystrophin negative fibers. Scale bar: 75 μm. In panels B and D serial sections are stained with haematoxylin and eosin staining to check tissue integrity and histopathology. Scale bar: 100 μm
Dystrophin immunoblottingIn all muscle biopsies, normal molecular weight but reduced amount of dystrophin was detected. Dystrophin levels were 49.2% ± 14.9% of those observed in healthy controls in the asymptomatic carriers, and 35.1% ± 28.6% in symptomatic patients (p = n.s.). No correlations between the amount of dystrophin, age at muscle biopsy (Fig. 3) and age at onset of muscular symptoms were observed.
Fig. 3
Scatter plot of dystrophin amount by age at muscle biopsy. Spearman’s rank test: rho = 0.34, p = 0.20
XCI pattern in blood DNAEighty-five percent (40/47) of participants were heterozygous at the AR locus, and the XCI pattern was assessed (Supplemental Table S4). Nine symptomatic DMD heterozygous females showed a random XCI pattern (9/18, 50%), and nine a skewed pattern (9/18, 50%). Among asymptomatic subjects, 8 showed random X inactivation (8/22, 36.5%), and 14 skewed X inactivation (14/22, 63.5%).
In six females, the segregation analysis of the DMD mutation in affected family members allowed for the identification of the X chromosome carrying the mutated DMD gene. This was possible when a DMD heterozygous female relative was available, and the phase between the AR and the DMD gene was clearly established. Among these six individuals, three were symptomatic, and in all the X inactivation favored the normal X chromosome, although the degree of inactivation was extremely variable, with only one individual showing a skewed XCI (#29) pattern, thus resulting in the severe symptomatic phenotype observed. The remaining three female carriers were asymptomatic with a random pattern of XCI.
From the ordinal regression analysis, the estimated effect of XCI on clinical severity showed a coefficient of 0.4505 suggesting that XCI does not predict clinical severity. Moreover, the Wald test did not indicate a statistically significant association (χ2 = 1.97, p = 0.16).
No significant correlation was observed between XCI pattern and cardiac involvement, nor skeletal muscle involvement (Fisher’s exact test p = n.s.).
Median CK values were 316.5 U/L in “random” and 400 U/L in “skewed” DMD heterozygous females (Wilcoxon rank-sum test p = 0.95, Supplemental Figure S3A).
The comparison of LV-EDVi between groups with random and skewed XCI (Supplemental Figure S3B) was assessed using a Wilcoxon rank-sum test. The median EDVi was 54 ml/m2 for the random and 50.5 ml/m2 for the skewed XCI groups (p = 0.55). Similarly, the comparison of LV-EF% between groups was not significant (p = 0.37), with median EF of 58% for the random XCI group and 60% for the skewed XCI group (Supplemental Figure S3C).
A Kaplan–Meier time-to-event analysis was performed to assess the median time of the onset of muscular symptoms with stratification based on the pattern of XCI (“skewed” vs. “random”). The median age at onset was 55 years for the random XCI group and 57 years for the skewed XCI group, with no statistically significant difference between the two groups (log-rank test, p = 0.38) (Fig. 4).
Fig. 4
Kaplan–Meier analysis comparing pattern of XCI (“random” vs. “skewed”) to age of onset of clinical manifestations in skeletal muscle of DMD heterozygous females.
The median percentage of XCI was 74.5% for the asymptomatic patients and 73.5% for the symptomatic ones, with no statistically significant differences between groups (Wilcoxon rank-sum test p = 0.6, Supplemental Figure S4).
Modifier genotypesDMD gene mutations were classified in deletions, duplications, nonsense, and intronic. The Fisher’s exact test revealed no statistically significant difference in the distribution of phenotypic severity across the various groups of DMD mutation types in our dataset (p = 0.56).
Genotype analysis of the modifier SNPs in SPP1, LTBP4 and CD40 genes was performed in 45 subjects (18 symptomatic patients and 27 asymptomatic carriers). We used a Cox regression model to estimate the effect of variants in SPP1, LTBP4 and CD40 genes on the onset of symptoms and a multiple linear regression model to evaluate the effect of the SNPs on quantitative factors (CK, LV-EDVi and LV-EF values). No correlation between the genotypes and any of the variables considered was observed.
Comments (0)