
Remember me



The overall primary objective of this project is to quantify patient preferences and relative importance of attributes for different device and administration features to deliver high-dose/high-volumes SC formulations of treatment, in people living with MS in the USA. This objective was derived from FDA feedback on a quantitative PPS originally planned to be conducted as a “global “ DCE study with patients from several countries (USA, France, Germany, China, Japan) and across a variety of diseases areas (Oncology, Asthma). The initial design of the study was based on a targeted literature review and patient and health care provider (HCP) input from an initial qualitative study. Its objective was then reviewed and adapted following FDA advice through consultation meetings (Table 1).
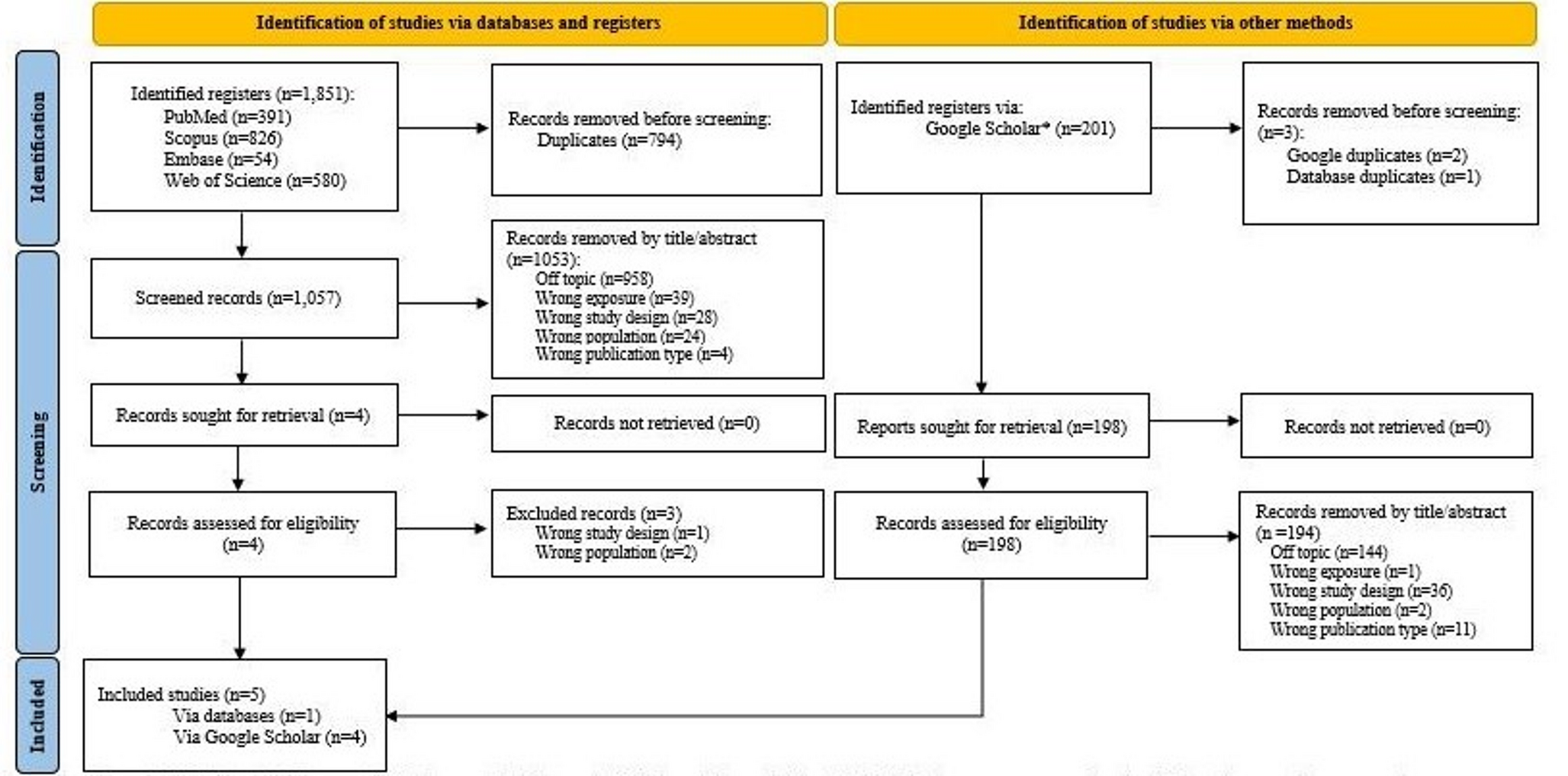
Table 1 Summary of the key FDA advice provided for the PPS design through informal and formal consultation meetings.This final study was designed following a staggered approach using three steps (a. Pre-FDA meetings: Qualitative research, b. FDA meetings and c. Post-FDA meetings: Qualitative research) which are summarized in the flow chart given in Fig. 1 and further explained in Sections A to C below. The planned DCE study is now more specific in that it aims to gain a thorough understanding of the device preferences in one country and in one indication but will not support answering the question whether one device type might be a preferred option across multiple indications, which would require additional studies.
Figure 1
Main stages of preparatory qualitative work.
This article focuses on how initial qualitative research informed the development of an A&L grid, what feedback from scientific advice meetings with the FDA was critical in the study design, why additional in-depth qualitative research was necessary, and the importance and benefit of collaboration with patient partners in a preference study.
The execution and reporting of the completed DCE will be conducted as a next phase and its results will be published in a separate article when available.
A. Pre-FDA Meetings: Qualitative ResearchInitial Qualitative Research Across Different Diseases and CountriesQualitative research was conducted in the USA and UK with 28 patients/caregivers and 20 HCPs to select and assess the importance of attributes related to SC injections. The questionnaire design for the qualitative research was informed through a prior targeted literature review based on patient experiences and needs regarding SC administration. This involved conducting telephone interviews with oncology, dermatology, respiratory and neurology patients and/or caregivers and with HCPs. The goal was to understand attributes of importance regarding the SC injection delivery, to gain insights into administration experiences, and elicit perspectives to hypothetical SC device concepts. The research sample recruitment by disease and specialty area is shown in Supplementary Material (SM) S1, and the discussion topics covered in the qualitative research are shown in SM S2. The respondents were asked to rate attributes related to SC injection by importance on a 0–10 numeric rating scale, with 0 being ‘not at all important’ and 10 being ‘most important’. Respondents were also asked to give their rationale for the ratings chosen.
The above findings were used to develop a “global” DCE study design presented to the FDA. The “global” study design originally included patients with different diseases (Oncology, Asthma) and different countries (USA, France, Germany, China, Japan).
B. FDA MeetingsNovartis sought scientific advice from the FDA-CDRH PPI group (through the Q-Submission Program) on the “global” DCE, with the overall process from first contact and briefing book preparation to receipt of the final advice taking approximately six months. Two informal teleconference meetings were held prior to the formal request for a pre-submission meeting. These meetings were key to understanding general requirements on the submission route and introduce the overarching concept of the sponsor’s PPS to the FDA, noteworthy because of the very early stage in the device development when this interaction was taking place (in advance of any drug-device combinations or considerations of benefit/risk trade-offs). The first informal meeting's objective was to obtain guidance on the most expeditious submission pathway and appropriate FDA division from whom to request guidance on the PPS. The potential unmet medical need and rationale for the PPS in the complex context of a device development for potentially different diseases, key aspects of study design, details of the PPS objectives and planned application of the results were presented. After expressing Novartis’ intention to seek advice through the PPI group’s dedicated email (CDRH-PPI@fda.hhs.gov), FDA provided the opportunity for the first informal teleconference. A second (follow-up) informal meeting with the FDA PPI team was set up to continue the first meeting's discussions. Those informal meetings helped Novartis to better understand the FDA’s thinking and the FDA’s responses were a key driver for the team to refine the design quite significantly.
The timing of milestones during the advice procedure with the FDA are shown in Table 2. The briefing book content was informed by PREFER and FDA pre-submission guidance [6, 25]. The document included an introduction, a list of seven questions, covering three substantial topics in line with the recommendations of the FDA Q-Sub Guidance on the number of topics and lastly, background information on the project (see SM S3). The topics of discussion at the meeting and the subsequent FDA advice were in-line with the aspects of the PPS framework identified as particularly relevant for discussion during the scientific advice process, as described in the PREFER recommendations [6]. The key questions which were addressed with the FDA covered the following three substantial topics of (1) appropriateness of study purpose and intended use of the study results, (2) appropriateness of study design (study objectives, attributes and levels, patient centric study materials, statistical methods, sample size, participants’ diagnosis) and (3) transferability of the results to additional disease areas.
Table 2 Duration and milestones for FDA consultation on a Patient Preference Study for a device (CDRH Q-submission route).C. Qualitative Research Post-FDA MeetingsBased on the discussions with FDA and their scientific advice, the design of the planned quantitative PPS was simplified to focus on one single disease (MS) and country (USA) instead of several indications across multiple countries. The updated study design allowed an increase in quality and robustness of the study results by focusing the study in one complex disease area (MS)/one diverse country and aligning the study population sample with the broader MS population including diversity in the MS patient demographics and characteristics. FDA recommended avoiding a broad scope of disease areas, since results may be too general and may have too many caveats to be useful in the end and may be more challenging to apply. The recommendation was to test the thinking in a specific area and then build from there to a broader scope. This required additional qualitative research to confirm the choice of attributes and levels in the PPS.
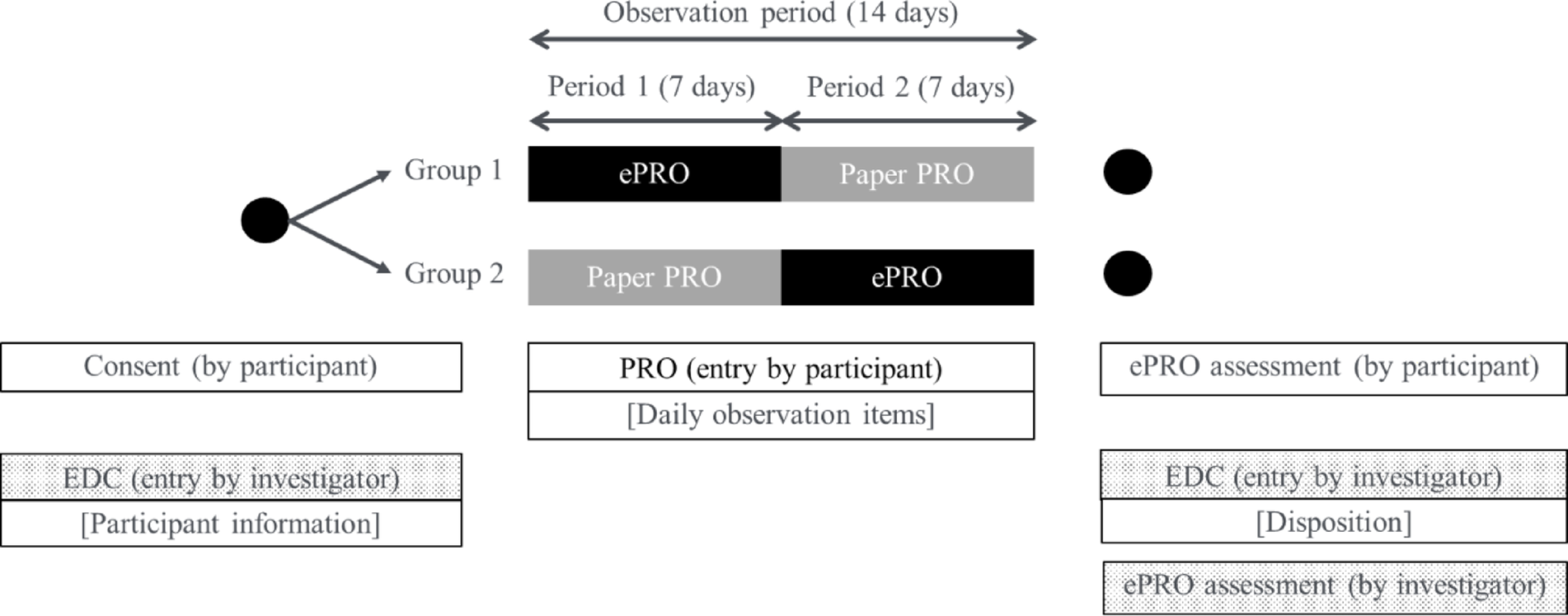
Additional in-Depth Qualitative ResearchAdditional qualitative research was conducted to address FDA’s feedback. Twenty-four USA-based MS patients took part in an in-depth interview via telephone or screen-sharing software (Microsoft Teams). A combined concept elicitation (CE) and cognitive debriefing (CD) approach was taken (Fig. 2). The interviews aimed to generate evidence to inform A&L development and ensure the future PPS is fit-for-purpose in an MS patient population.
Figure 2
Structure of the concept elicitation and cognitive debriefing interviews.
Concepts spontaneously discussed by MS patients were subject to concept saturation by the research team (see SM S4). Concept saturation is defined as ‘the point at which no new concept-relevant information emerges’ [26]. Concept saturation strengthens the study design by supporting the notion that enough interviews were conducted to be sufficient to capture the key treatment drivers of interest to MS patients. The FDA Patient-Focused Drug Development Series (specifically, FDA PFDD Guidance 2) [27] outlines guidance and agency expectations on using concept saturation to justify qualitative sample sizes. The interviews included participants across MS subtypes: Relapsing Remitting MS (RRMS), Secondary Progressive MS (SPMS), and Primary Progressive MS (PPMS). The sample size for each MS subtype was in accordance with concept saturation principles [28].
Each interview lasted approximately 60 min and was conducted by trained, experienced moderators who were briefed on population-specific considerations. Interviews were transcribed verbatim and subject to framework and thematic analysis methods [29,30,31]. An induction–abduction approach was taken to identify themes emerging directly from the data (inductive inference), and by applying prior knowledge (abductive inference).
The CE section involved asking broad, open-ended questions to encourage MS patients to discuss their treatment experiences and highlight features that are important when evaluating MS injection treatments.
The CD section used a ‘think-aloud’ approach [27, 32, 33] to assess consistency of understanding and interpretation, assess the relevance and importance of A&Ls selected, and confirm that appropriate framing (language and images) was used. The levels for each attribute were also explored to confirm if level differences were salient and differentiating. MS patients were asked to read aloud and discuss every A&L while responding to questions from the interviewer.
Study exemptions and approvals for the qualitative studies pre- and post-FDA meetings were granted by a centralized Institutional Review Board in the USA.
A to C Involvement of Patient PartnersThe research team worked in collaboration with patient partners on the research work pre- and post- FDA meetings as well as to prepare the Scientific Advice meeting with the FDA.
The patient partners were selected and were adapted as the project progressed to represent the target population in the different study designs. The rationale for involvement of patient panels for the “global” DCE study design pre-FDA consultation and MS DCE study design post-FDA consultation is described in Table 3. Patient partners were selected by the sponsor’s internal Patient Advocacy group in contact with patient organizations and communities. Key criteria for selecting patient partners included representativeness of the targeted disease population, ensuring a range of patient experiences, considering geographical representation, targeted characteristics like age or gender, cultural considerations, and English fluency (Table 3). Each patient partner was contracted individually and compensated for the time spent on the study, using the Novartis version of the Fair Market Value calculator developed by the National Health Council [34]. Individual on-boarding sessions with the patient partners were performed by the Patient Engagement representative and Project Lead to ensure sound understanding of the objectives of the PPS as well as their role as patient partners and related expectations. In addition, the Project Lead and Patient Engagement representative provided, during these on-boarding sessions, the objectives of the project beyond the PPS, the different activities performed in its scope and how the PPS will be used to inform this project. Interactions with the patient partners were conducted individually and virtually, at each key milestone of the study to collect their feedback, discuss and clarify any questions. Based on their feedback, the draft PPS documents were revised as required.
Table 3 Panel of patient partners prior to and following the FDA scientific advice consultation.Patient partners helped to develop and review study material from its conception. The patient partners reviewed and provided comments on the concept sheet, giving an overview of the study design and device features, treatment process, attributes, and levels to be tested. The patient partners were then asked to review and comment on additional documents: the study protocol, quantitative survey script, Informed Consent Form, and educational materials to be provided to study participants (videos and infographics, as built into the study survey; see SM S5). Later, the patient partners were involved in reviewing and refining a test version of the online quantitative survey. The study team did not provide any guidance or specific direction as to which sections of the documents should be reviewed. Nevertheless, the Patient Engagement representative and Project Lead provided the PPS context in the project and were always available to connect with the patient partners, to ensure a clear understanding of the review expectations. Patient partners were also involved in reviewing the outputs of the initial qualitative research and FDA scientific advice feedback and providing context for the interpretation of the study results. Patients were not given a target number of hours to review the different documents. However, they were requested to carefully track the time spent on the review, for compliance and financial reasons. As an example, review of the draft protocol took a maximum of 10 h for a patient partner.
The feedback and comments from the above were used to develop the “global” DCE study design that was presented to the FDA as well as the DCE study design refined after the Scientific Advice with FDA. Following the FDA scientific advice, and the restriction of the PPS scope to MS patients and USA only, additional patient partners, who were MS patients living in the USA, were asked to review the revised study material.
Comments (0)