
Remember me



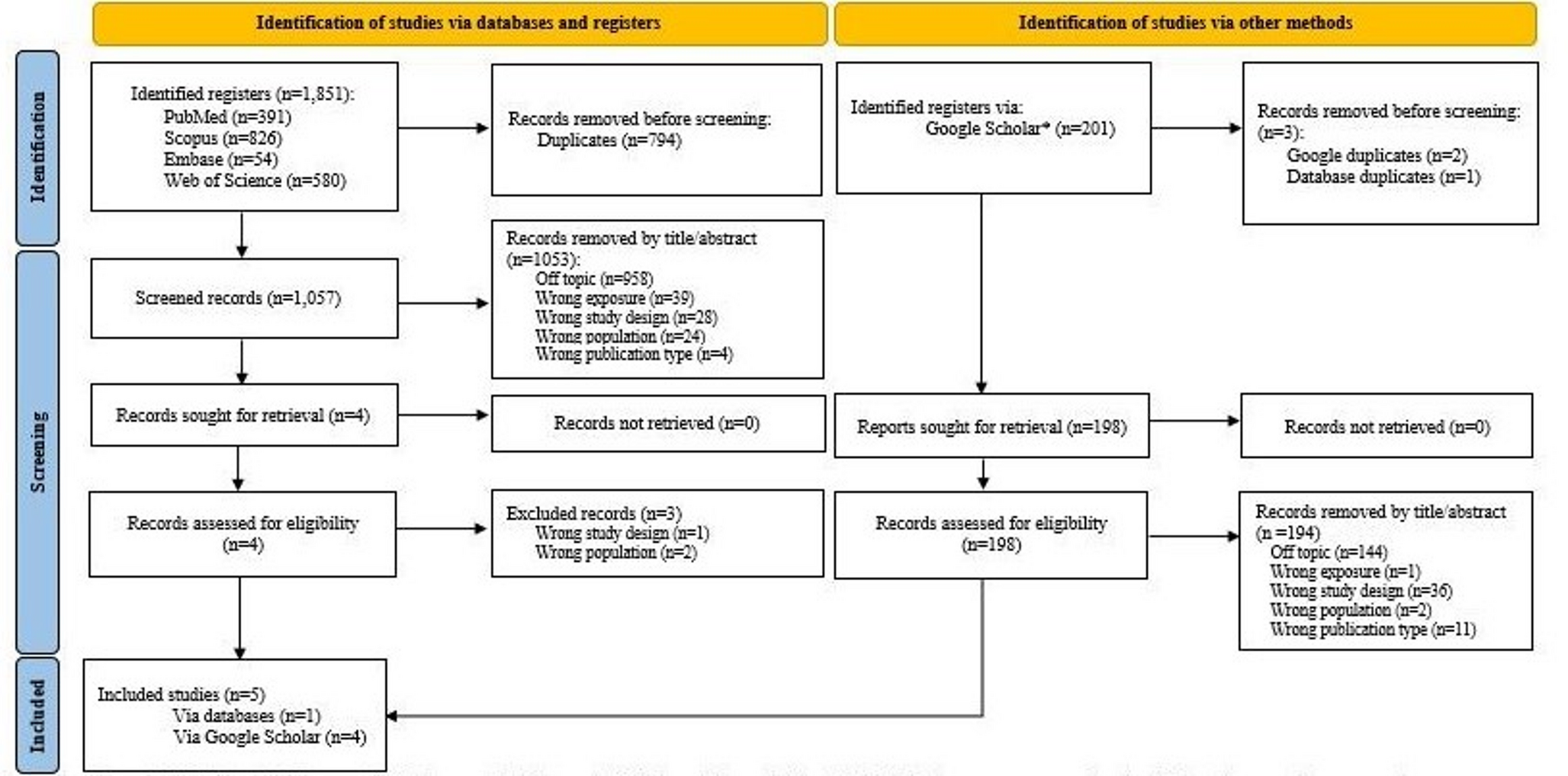
Regulatory guidance documents on safety in pregnancy include four main areas for consideration: Product development, aspects relevant to interventional studies, requirements relevant to the post-marketing setting, and elements applicable to the full lifecycle (Fig. 2). These areas were addressed in separate sections of the TransCelerate “Points to Consider Concerning the Use of Medicines in Pregnancy throughout the Product Lifecycle (Based on the Regulatory Guidance across the Globe)” report document [28]; the product development section of this report document addresses considerations for how and when to include pregnant women in the drug development process [28]. The interventional study considerations include details regarding international regulatory guidelines relating to enrollment in clinical trials and follow-up for clinical trials. Post-marketing study setting considerations include regulatory and international guidelines’ requirements for case reports, risk management, and non-interventional studies. The full lifecycle considerations section includes important aspects related to signal detection, aggregate reports, and labelling regulations.
Fig. 2
Areas of consideration from regulatory guidance documents on safety in pregnancy comprise of four main areas: product development, aspects relevant to interventional studies, requirements relevant to the post-marketing setting, and elements applicable to the full lifecycle
Product Development ConsiderationsDisease ConsiderationsGuidance on pediatric studies from both the US iPSP and the EU PIP includes disease presentation as an important consideration that may be applicable to the evaluation of medicines in pregnancy. Disease presentation includes a description of the etiology of the disease / condition, clinical manifestations, prognosis, prevalence, incidence, and epidemiology [18, 25]. Disease considerations include the context of current medical practice and available therapies [30]. Similar to pediatrics, if the disease is not prevalent in WOCBP, the development and post-marketing plans would differ from a disease that has a high prevalence in those populations.
Medication CharacteristicsMedication characteristics such as the indication, mechanism of action, pharmacokinetics (PK), pharmacodynamics, sites of action, and potential or expected side effects are important considerations in both the EU and US.
The potential differences in the metabolic and PK aspects for pregnant women must be considered and compared to the general population. The US guidance on inclusion of pregnant women in clinical trials also highlights the importance of understanding the PK of a molecule. Because of the extensive physiological changes associated with pregnancy, PK parameters may change, sometimes enough to justify changes in dose or dosing regimen. Therefore, FDA guidance provides that PK data in pregnant women should be collected during clinical trials to guide appropriate dosing [8].
Non-clinical ConsiderationsAs mentioned in the US and EU pediatric guidance, findings from animal and reproductive toxicology studies are important in developing a medication. It is necessary to determine whether prerequisites to human, maternal administration exist [18, 25]. Appropriate animal models and reproductive toxicology studies should be considered when developing a medication for pregnant women [18, 25]. It is important to identify what is known from animal reproductive toxicology studies and pregnancy models and whether a medication affects the growth and/or maturation of the fetus. The relevance to humans from these findings should be considered.
Potential teratogenic and genotoxic characteristics of the medication should be considered. Global guidelines advise performing female reproduction toxicity studies and standard genotoxicity tests before testing a medication in pregnant women in clinical trials. Non-clinical safety studies should be performed before human clinical trials and marketing authorization for pharmaceuticals (ICH guideline M3(R2)) [31].
Clinical ConsiderationsSimilar to guidance in the pediatric development programs, there is a need for an improved benefit risk profile in a new molecular entity being developed in the population as compared to the current standard of care of the condition of interest [9, 18, 25]. It is critical to take into account the expected benefit, expected improvement in safety profile, including adverse event profile, drug interactions, potential medication errors, and the availability of clinically relevant and new therapeutic knowledge for the use of the medicinal product in the population [18, 25]. These should also be taken into consideration when determining medications for clinical development in pregnant women.
There may be challenges in performing clinical trials with pregnant women and WOCBP depending on the medication. Therefore, it is critical to reflect upon the feasibility of studying the population and condition of interest in both the clinical and post-marketing settings [18].
There are additional considerations for a new drug or new indication, where there is anticipated or actual use of the drug in pregnant women. Research involving pregnant women should demonstrate potential health benefits to pregnant women or the fetus, and new therapies would be expected to improve pregnancy and/or fetal outcomes as compared to the existing therapy. Any potential benefit to the mother and the fetus should be weighed against possible risks to both the fetus and the pregnant woman.
Feasibility of performing clinical trials investigating the condition of interest should be deliberated. The data needed to support the design or initiation of studies in paediatrics in addition to what may be needed for a feasibility assessment are critical factors [11]. These considerations would also apply to studies in women of childbearing potential and pregnant women.
Interventional Study ConsiderationsEnrollment in Clinical TrialsCurrent regulations acknowledge that enrollment of pregnant women in clinical trials is complicated as exposure to an investigational product may present risks to the mother and fetus. Findings from non-clinical and animal studies should inform initial human doses and duration of exposure [9].
For WOCBP included in clinical trials, ICH M3 R2 includes additional guidance to minimize and characterize the risk of unintentional exposure of the pregnant woman, thus of the embryo and fetus. Canadian guidelines further clarify that enrollment decisions should be based on careful risk-benefit evaluation and consider factors such as “the nature and severity of the disease, the availability and results of previous nonclinical data on pregnant and non-pregnant animals, and results from clinical data” [30].
Follow-up for Clinical TrialsAccording to ICH E8 [32] and CIOMS VI [33], if a pregnancy occurs during a clinical trial, follow-up should be conducted for the health of the fetus and the pregnancy outcome (e.g., pregnancy termination, type of birth). Monitoring the pregnancy of a woman whose male partner is a trial participant may be needed in certain situations, such as when there is evidence of a class effect or when there is new evidence from animal models [33]. The duration of the follow-up is not clearly specified in the regulatory guidelines. The Canadian Guidance Document [30] and CIOMS VI [33] provides that, when possible, monitoring the development of the newborn and long-term follow-up of a child should be considered.
Post-Marketing Setting ConsiderationsCase ReportsRegarding case reports, the US, EU, Canadian, Australian, Singapore, South African health authorities, and ICH all require MAHs to collect as much pregnancy exposure information as possible.
Requirements differ when considering which cases qualify for expedited reporting or which cases need only be discussed in periodic safety update reports. However, regulations indicate pregnancy exposure information should be collected and discussed in the periodic safety update reports [34,35,36,37,38,39]. In general, ICH, US, EU, Canadian, and the Australian guidelines provide that all reports where the embryo or fetus may have been exposed to medicinal products should be followed up [35, 40].
Risk ManagementPer the EU guidance, “additional pharmacovigilance (PV) activities in the risk management plan (RMP) should be taken in a risk proportionate manner, considerations regarding risk proportionality will differ for the populations of pregnant women and breastfeeding women. The objective of the risk mitigation measure (RMM) is to reduce any risk to the child as much as possible given the need for treatment in the mother.” For products with anticipated use in WOCBP, a reflection of current understanding of safety in pregnancy should be included in the summary of the safety specifications in the RMP. The RMP should specifically discuss the likelihood of the use of the medicine in pregnancy, and WOCBP considering the proposed indications, alternative treatment options, need for effective contraception, and complexities of changing treatment if use during pregnancy is to be avoided [41, 42].
Non-Interventional StudiesMost information on the outcomes of drug exposure in pregnant individuals still relies on data generated post-approval. The ICH and CIOMS recognize that additional studies in the post-market setting may be warranted for pregnant women [43, 44]. In most regions reviewed, there were no clear regulations or guidance on when to consider these post-marketing studies nor best practices for conducting them. In regions where regulatory guidance for post-authorization pregnancy safety studies exists (primarily US and EU), recommendations tended to diverge from one another, with the level of detail included varying significantly between countries/regions.
The US guidance includes recommendations for when a product developer should establish a pregnancy exposure registry [37]. EU guidance provides that epidemiological studies should be carried out using existing data sources (i.e., secondary data) and be designed in such a way as to minimize bias [45]. In contrast, the US generally considers secondary data sources (e.g., electronic data sources, such as insurance claims and electronic health record databases) as additional studies that complement data obtained from pregnancy registries [39].
Full Lifecycle ConsiderationsSignal DetectionICH [43], CIOMS [46], Australia [40], EU [38, 41, 47], Eurasian Economic Union (EAEU) [43], Saudi Arabia [31], Switzerland [48], and US [37] guidance and regulations mention signal detection in pregnant women. As per CIOMS [46] and EU [48] guidance and regulations, the methods for signal detection depend upon the product, indication, type of adverse outcome to be monitored, and magnitude of risk. In addition, possible signals of teratogenic effects are a significant safety issue and, per regulations in Australia, EU [35], Saudi Arabia [31], and Switzerland [49], may require timely notification to health authorities.
Aggregate ReportsPer ICH, EU [38, 41, 47] and India [49], pregnant or lactating women exposed during clinical trials or post-approval should be included as a special exposure population in both the development safety update report (DSUR) and periodic safety update report in the periodic benefit risk evaluation report (PBRER), also referred to as the periodic safety update report (PSUR). Whereas ICH E2C (R2) does not require a summary of pregnancy outcomes in the PBRER, Australia and EU do require a summary of pregnancy outcomes to be included in the PSUR.
Labelling RegulationsInternational labelling provisions for pregnant or lactating women are similar, but country-specific requirements can vary. In 2008, Europe implemented guidance on the risk assessment of medicinal products on human reproduction and lactation, entitled ‘From data to labelling’ [50]. The FDA initiated the Pregnancy Lactation Labeling Rule (PLLR), effective June 30, 2015, which includes descriptive language on adverse outcomes following drug exposure and specific disease states during pregnancy [51].
The FDA requires manufacturers to update their Prescribing Information (PI) when new information becomes available that causes the current PI to be inaccurate, false, or misleading [52]. Updates may include indications, uses, populations, dosages, safety information, or other information on how to use the medicine safely and effectively [52].
Comments (0)