
Remember me
Pregnancy-induced hypertension (PIH) is a pregnancy-specific condition characterized by increased blood pressure, proteinuria, and edema and often results in damage to multiple organs, thus posing a significant risk to maternal and fetal health. PIH remains a major contributor to maternal and perinatal mortality.[1,2] Hypertension leads to vascular endothelial damage.[3] Endothelial injury is associated with decreased nitric oxide availability, which ultimately initiates and exacerbates various vascular diseases.[4,5] The nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome is a protein complex in the NOD-like receptor family that features a pyrin domain and a heat shock protein domain and is closely associated with the onset of inflammatory disorders. When the NLRP3 inflammasome is activated, cysteinyl aspartate-specific proteinase 1 (caspase-1), a cysteinyl aspartate-specific protease, cleaves pro-interleukin-1β (IL-1β), pro-interleukin-18 (IL-18); and gasdermin D (GSDMD) to induce the maturation of IL-1β, IL-18, and the N-terminal fragment of GSDMD (GSDMD-N). This sequence of events initiates a specific form of inflammatory cell death known as pyroptosis.[6-8] During NLRP3 activation, damaged endothelial cells secrete various inflammatory mediators.[9] Furthermore, sustained NLRP3 activation accelerates pyroptosis, resulting in significant endothelial cell loss and subsequent endothelial dysfunction.[10] Inhibiting pyroptosis in endothelial cells enhances their survival, promotes angiogenesis, and improves the prognosis of ischemic diseases.[11] Thus, blocking NLRP3 activation may provide a promising approach to manage inflammatory conditions associated with pyroptosis.
NLRP3 activators, such as adenosine 5'-triphosphate (ATP) or streptolysin, induce mitochondrial dysfunction and lead to NLRP3 activation by decreasing mitochondrial membrane potential (MMP) and releasing mitochondrial damage-associated molecular patterns, including mitochondrial reactive oxygen species (ROS).[12-14] Therefore, mitochondrial dysfunction is crucial in triggering NLRP3 activation through multiple pathways.[15,16] The mitochondrial ROS scavenger mito-TEMPO can inhibit NLRP3 activation in cells,[17] suggesting a direct link between the oxidative stress from mitochondrial dysfunction and the activation of NLRP3 inflammasome during inflammation. Mitochondrial autophagy has gained increasing attention for its role as a critical cellular self-repair mechanism in various diseases.[18,19] It facilitates the removal of damaged mitochondria, making it crucial in maintaining mitochondrial balance and ensuring mitochondrial quality control.[20]
The binding of pro-IL-1α to mitochondrial cardiolipin inhibits the interaction between microtubule-associated protein 1 light chain 3B and mitochondria, preventing mitochondrial autophagy and leading to the accumulation of damaged mitochondria. This accumulation further stimulates NLRP3 activation and exacerbates IL-1β release.[21] Therefore, the pathological mechanisms of mitochondrial dysfunction, oxidative stress, and impaired mitochondrial autophagy collectively accelerate NLRP3 activation. Maintaining mitochondrial homeostasis and enhancing mitochondrial autophagy to suppress NLRP3 activation and subsequent pyroptosis may provide a viable strategy for treating inflammatory diseases.
The aim of this study is to investigate the role of mitophagy in mitigating vascular endothelial cell damage in PIH. Specifically, we seek to explore how mitophagy may influence the suppression of NLRP3 inflammasome activation and its subsequent impact on pyroptosis in endothelial cells. By elucidating the mechanisms through which mitophagy regulates mitochondrial homeostasis, we aim to provide deeper insights into its potential therapeutic effects on vascular injury in PIH.
MATERIAL AND METHODS Cell cultureA human umbilical vein endothelial cell (HUVEC) line (PCS-100-010) was obtained from the American Type Culture Collection (Manassas, VA, USA). All cells were authenticated through short tandem repeat profiling, and mycoplasma testing was conducted to prevent contamination. The cells were cultured individually in their respective culture media. The HUVECs were maintained in a HUVEC complete medium (10% fetal bovine serum, 100 U/mL penicillin-streptomycin, 1% endothelial cell culture supplement) (iCellh110-001b, iCell-h110, Shanghai, China), seeded into 25 cm2 culture flasks, and incubated in a 37°C, 5% carbon dioxide (CO2) incubator (BPN-80CH, Shanghai, China). Subculture was performed every 3–4 days based on cell growth, and cells were selected for experimental studies as necessary. The experimental groups were as follows: Control group, L-NAME group (PIH group, induced with 300 µM L-NAME for 24 h following the method of Chen et al.),[22] L-NAME + 3-methyladenine (3-MA; HY-19312, MedChemExpress, New Jersey, USA) group (hypertension group treated with 5 mM 3-MA for 2 h to inhibit mitochondrial autophagy), and L-NAME + rapamycin (Rapa; HY-10219, MedChemExpress, New Jersey, USA) group (hypertension group treated with 100 nM Rapa for 4 h to activate mitophagy).
Cell viability assayCell viability was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The treated HUVECs were seeded in 96-well plates (5 × 104 well/mL) and cultured until they reached the required confluence, after which they were treated under the respective conditions.
After treatments, the cells were added with the MTT (G4101, Servicebio, Wuhan, China) reagent and incubated for 4 h. Dimethyl sulfoxide (ST1276, Beyotime, Shanghai, China) was subsequently incorporated to dissolve the formed purple-blue formazan crystals. Absorbance at 490 nm was measured using a multifunctional microplate reader (CMaxplus, MOLECULAR DEVICES, USA) to determine cell viability. Each condition was evaluated in triplicate, and the experiment was performed at least 3 times.
Lactate dehydrogenase (LDH) assayFollowing treatment as per the experimental groups, the cell supernatants were collected and centrifuged at 3,000 rpm for 20 min at 4°C. The concentration of LDH (A020-2, Nanjing Jiancheng, Nanjing, China) in the supernatant was quantified in accordance with the guidelines provided by the manufacturer of the enzyme-linked immunosorbent assay kit.
ROS assayThe treated cells were collected, and the culture medium was discarded. Then, 500 µL of diluted 10 µM 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) (S0033S-1, Beyotime, Shanghai, China) solution was added to ensure full cell exposure. The cells were incubated at 37°C in an atmosphere containing 5% CO2 for 20 min. After incubation, the cells were thoroughly rinsed with serum-free culture medium 3 times to remove any unabsorbed DCFH-DA. The cells were then observed and photographed under a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan). Analysis and quantification were performed using ImageJ software (V1.46; National Institutes of Health, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) stainingThe cells were seeded into 24-well plates (5 × 104 well/mL) and cultured overnight, followed by apoptosis induction and two washes with phosphate-buffered saline (PBS) (G4202, Servicebio, Wuhan, China). The cells were then fixed with 4% paraformaldehyde at 4°C for 25 min, followed by two washes with PBS. The cells were permeabilized with a 0.2% Triton X-100 (T8200, Solarbio, Beijing, China) solution. After three washes with PBS, the cells were added with the labeling solution (K1133, APExBIO, Shanghai, China) and incubated in the dark for 60 min, followed by additional washes with PBS. The cells were stained with 4',6-diamidino-2-phenylindole (DAPI) (C0060, Solarbio, Beijing, China) and washed 3 times with PBS. An anti-fade reagent (S2100, Solarbio, Beijing, China) was added, and images were captured using a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan).
MMP detectionStaining buffer and working solutions were prepared according to the instructions of the 5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) assay kit (C2006, Beyotime, Shanghai, China). In brief, a six-well plate was added with 1 mL of working solution each well, incubated for 20 min, and then observed and photographed using a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan).
ImmunofluorescenceThe cells were cultured on coverslips placed in six-well plates (5 × 103 well/mL), fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% goat serum. Primary antibodies against microtubule-associated protein 1 light chain 3 (LC3) (14600-1-AP, Proteintech, Wuhan, China) and GSDMD (HA601046, HUABIO, Hangzhou, China) were applied, and the cells were incubated overnight at 4°C, followed by washing and incubation with the appropriate secondary antibodies (GB22303, Servicebio, Wuhan, China). After additional washes, the cells were stained with DAPI for 10 min. Finally, fluorescence images were captured using a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan). Image analysis and quantification were performed using ImageJ software (V1.46, National Institutes of Health, USA).
Western blot (WB) analysisFollowing treatment, the cells were washed 3 times with PBS and lysed on ice with radio immunoprecipitation assay buffer (P0013C, Beyotime, Shanghai, China) for 30 min to extract total protein. The proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (P0015A, Beyotime, Shanghai, China) and transferred onto a polyvinylidene fluoride membrane. The membrane was blocked for 2 h and then incubated overnight at 4°C with the primary antibodies against LC3 (1:1,000 dilution, 14600-1-AP, Proteintech, Wuhan, China), sequestosome 1 (P62) (1:1,000 dilution, 80294-1-RR, Proteintech, Wuhan, China), NLRP3 (1:1,000 dilution, 68102-1-lg, Proteintech, Wuhan, China), caspase-1 (1:1,000 dilution, ET1608-69, HUABIO, Hangzhou, China), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1,000 dilution, TA-08, ZSGB-BIO, Beijing, China). The membrane was then incubated with the appropriate secondary antibodies (1:2000 dilution, ZB-2305, ZSGB-BIO, Beijing, China) at room temperature for 2 h. Chemiluminescence detection was performed using the enhanced chemiluminescence reagent (Tanon-4600, Yuanping Hao Biotechnology, Beijing, China), and images were acquired using a gel imaging system. Protein band intensities were quantified using ImageJ software (V1.46, National Institutes of Health, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)After treatment, total RNA was extracted from the cells using the TRNzol Universal RNA Extraction Reagent (DP424, TIANGEN Biotech, Beijing, China). The extracted RNA was subsequently reverse transcribed into complementary DNA using the reverse transcription kit (KR116, TIANGEN, Beijing, China). PCR amplification was performed using specific forward and reverse primers on a real-time fluorescence quantitative PCR system (LightCycler96, Roche, Switzerland), with GAPDH as the internal control.[22] The relative gene expression levels were quantified using the 2−△△CT method. The sequences of the primers are provided in Table 1.
Table 1: Primer information.
Gene Primer sequences (5'-3') LC3 R: GCTCGTAGATGTCCGCGATThe small interfering RNA (siRNA) targeting the NLRP3 gene (siNLRP3) and the control siRNA (nontargeting control [siNC]) used for transfection were designed and synthesized by Jiangsu Kaiji Biotechnology Co., Ltd. (China). The siNLRP3 sequences were 5'-GUAUGAGAGUAGAGCGAUUUU-3' and 5'-GCUAAUGAUCGACUUCAAU-3', and the siNC (negative control) sequences were 5'-UUCUCCGAACGUGUCACGUTT-3' and 5'-ACGUGACACGUUCGGAGAATT-3'. HUVECs in the logarithmic growth phase were seeded in six-well plates and cultured until they reached approximately 80% confluence. The cells were then washed with PBS once and transferred to a serum-free medium. In accordance with the manufacturer’s protocol for Lipofectamine 2000 (11668027, Thermo Fisher Scientific, Waltham, MA, USA), the siRNA was mixed with the transfection reagent and applied to the cells, which were then incubated for 48 h. After incubation, the transfected cells were harvested and assigned to different experimental groups as follows: L-NAME + siNC (siNC), L-NAME + siNLRP3 (siNLRP3), L-NAME + siNLRP3 + 3-MA (siNLRP3 + 3-MA), and L-NAME + siNLRP3 + Rapa (siNLRP3 + Rapa). Subsequent experiments were performed as previously described.
Statistical analysisStatistical analysis was conducted using GraphPad Prism software (version 9.5, GraphPad Software, La Jolla, CA, USA). Data for normally distributed quantitative variables were expressed as the mean ± standard deviation. Comparisons among multiple groups were performed using one-way analysis of variance, followed by Bonferroni’s post hoc test. Pairwise comparisons were conducted using an independent sample t-test. Comparisons between two groups were analyzed using the t-test. Post hoc analysis was performed using the Tukey method. A P < 0.05 was considered statistically significant.
RESULTS Effect of mitophagy on the viability of HUVECs and the expression of autophagy-related proteinsThe role of mitophagy in L-NAME-induced vascular endothelial cell injury was investigated, and the following experimental results were obtained:
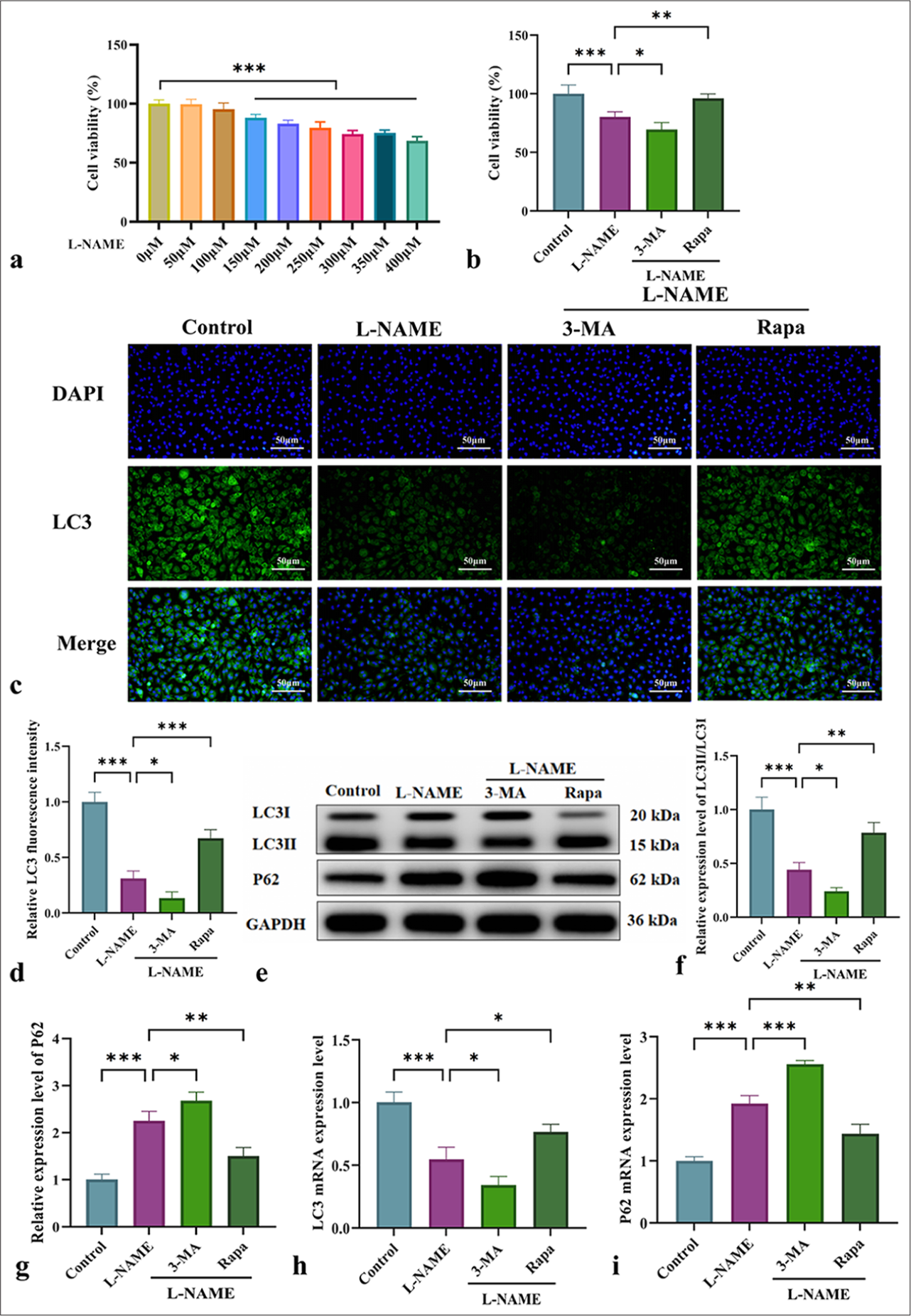
Figure 1a shows that exposure to 300 µM L-NAME for 24 h resulted in a significant decrease in cell viability. MTT assay indicated that L-NAME significantly inhibited cell activity, and 3-MA further exacerbated this effect. Meanwhile, Rapa significantly enhanced cell viability compared with L-NAME alone [Figure 1b] (P < 0.01). As shown in Figure 1c and d, immunofluorescence analysis of LC3II/LC3I expression demonstrated that L-NAME markedly reduced fluorescence intensity, which was further exacerbated by 3-MA. By contrast, Rapa significantly increased fluorescence intensity. WB analysis of LC3II/LC3I and P62 protein expression [Figure 1e-g] revealed that L-NAME significantly downregulated LC3II/LC3I while upregulating P62, and 3-MA further exacerbated these effects. By contrast, Rapa significantly upregulated LC3II/LC3I (P < 0.01) and downregulated P62 (P < 0.01) [Figure 1e-g]. These findings were consistent with the immunofluorescence results, as similar trends were observed for LC3 and P62 expression at the messenger RNA level [Figure 1h and i]. In conclusion, mitophagy plays a crucial protective role in L-NAME-induced vascular endothelial cell injury.
Export to PPT
Effect of mitophagy regulation on HUVEC damage and oxidative stressThe effects of different treatments on cell damage and oxidative stress were compared to investigate the role of mitophagy in L-NAME-induced endothelial cell injury.
TUNEL staining [Figure 2a and b] revealed a significant increase in fluorescence intensity after L-NAME treatment, indicating cell damage. This increase was further enhanced by 3-MA but significantly diminished by Rapa (P < 0.001). Measurement of LDH levels in HUVECs across different groups revealed that L-NAME significantly elevated the LDH levels, indicating cellular damage. 3-MA further increased the LDH levels, and Rapa significantly decreased them [Figure 2c] (P < 0.05). This finding was consistent with the TUNEL staining results. ROS fluorescence analysis was performed to assess the effect of L-NAME-induced oxidative stress. Figure 2d and e shows that L-NAME markedly elevated the ROS levels, indicating that it induces oxidative stress and endothelial cell damage. 3-MA further elevated the ROS levels, and Rapa significantly reduced them (P < 0.01). These findings suggested that mitophagy exerts a protective effect against L-NAME-induced endothelial cell damage by modulating oxidative stress and regulating cell death pathways.

Export to PPT
JC-1 dye staining was performed to assess MMP in HUVECs to further investigate the protective role of mitophagy in cellular injury [Figure 2f and g]. The control group displayed a high MMP, characterized by strong red fluorescence. After L-NAME treatment, the MMP decreased, indicating mitochondrial dysfunction. 3-MA exacerbated the damage, and Rapa promoted mitophagy, restored red fluorescence, and alleviated L-NAME-induced injury. These results indicated that mitophagy plays a pivotal role in endothelial cell injury associated with gestational hypertension. Its activation can suppress NLRP3-mediated pyroptosis, thereby mitigating cellular damage.
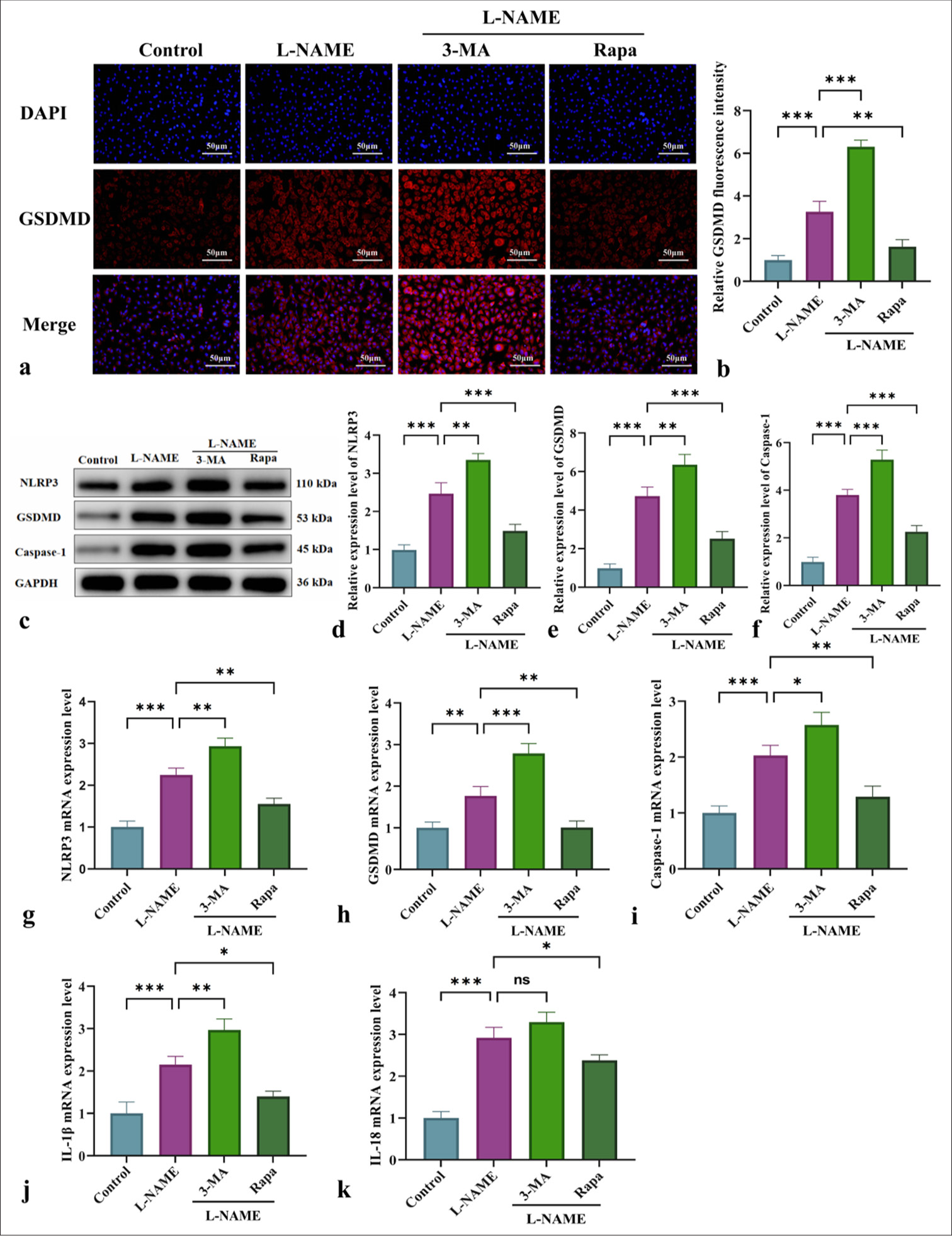
Effect of mitophagy on NLRP3-mediated pyroptosis and inflammatory marker regulationThe involvement of NLRP3-induced pyroptosis in endothelial cell damage following L-NAME treatment was investigated by evaluating the effects of different interventions on the expression of NLRP3, GSDMD, caspase-1, and pro-inflammatory factors (IL-1β and IL-18). Immunofluorescence, WB, and qRT-PCR analyses [Figure 3] demonstrated that L-NAME significantly activated the NLRP3 inflammasome, enhanced GSDMD cleavage, and promoted caspase-1 activation, thus initiating pyroptosis and the secretion of inflammatory mediators such as IL-1β and IL-18 [Figure 3]. Following 3-MA intervention, pyroptosis was further increased, accompanied by enhancements in NLRP3 inflammasome activation and GSDMD cleavage [Figure 3]. Conversely, Rapa alleviated NLRP3-mediated pyroptosis by activating mitophagy, thus significantly reducing the inflammatory response [Figure 3] (P < 0.05 or P < 0.001). In summary, mitophagy may serve as a potential therapeutic target for mitigating vascular endothelial damage associated with conditions such as gestational hypertension by modulating pyroptosis and NLRP3 inflammasome activation.
Export to PPT
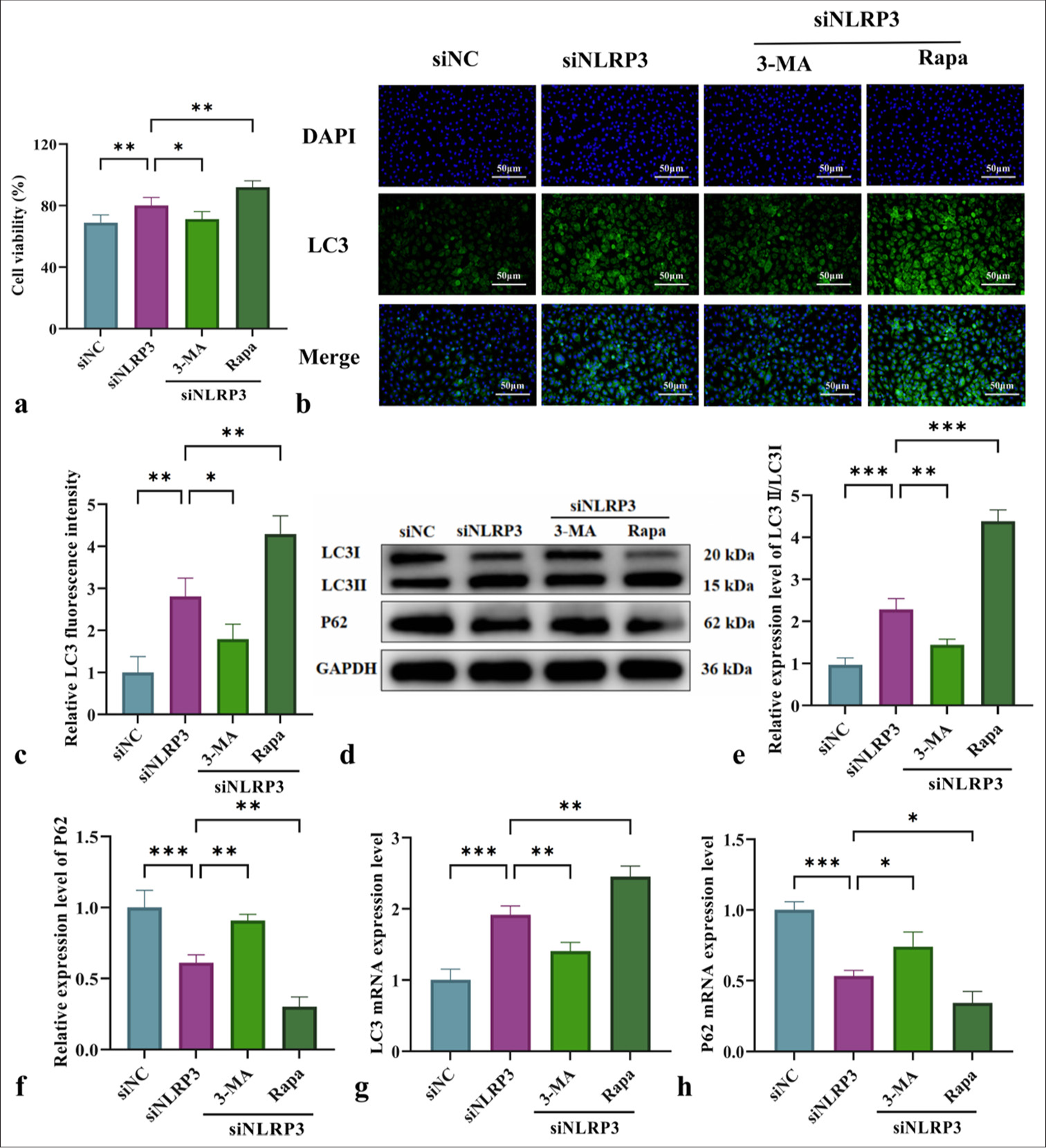
Effect of NLRP3 silencing combined with mitophagy regulation on the cell viability and autophagy levels of L-NAME-treated HUVECsThe impact of NLRP3 silencing on the cell viability and autophagy levels of L-NAME-treated HUVECs was assessed by interfering with NLRP3 expression and using mitochondrial autophagy-related drugs 3-MA and Rapa. MTT assay results in Figure 4a demonstrated that NLRP3 silencing reduced cell viability following L-NAME treatment, with a further decline observed after 3-MA intervention. Conversely, Rapa significantly enhanced cell viability. Immunofluorescence analysis revealed a significant increase in LC3II/LC3I fluorescence intensity in the NLRP3-silenced (siNLRP3) group. However, autophagy activation was suppressed by 3-MA, resulting in a reduction in fluorescence intensity. By contrast, Rapa markedly increased autophagy fluorescence intensity [Figure 4b and c]. WB and qRT-PCR analyses further confirmed the trends in LC3II/LC3I and P62 expression patterns, indicating that silencing NLRP3 improved L-NAME-induced cell damage by regulating mitophagy [Figure 4d-h]. The most significant effect was observed with Rapa intervention [Figure 4d-h] (P < 0.01). In summary, mitophagy plays a critical protective role in mitigating NLRP3-mediated cell damage.
Export to PPT
Effect of NLRP3 silencing combined with mitophagy regulation on L-NAME-induced cell damage and oxidative stress in HUVECsThe role of NLRP3 silencing and mitochondrial autophagy regulation on L-NAME-induced vascular endothelial cell injury was assessed across different treatment groups. As shown in Figure 5, L-NAME significantly increased the LDH levels, ROS production, and number of TUNEL-positive cells, indicating aggravated cellular injury. Figure 5 demonstrates that NLRP3 silencing (siNLRP3 group) partially alleviated these injuries. Further modulation of mitochondrial autophagy, either through inhibition by 3-MA or activation by Rapa, exerted differential regulatory effects on cellular injury. 3-MA aggravated cell damage, and Rapa effectively alleviated these injuries, showing a significant regulatory effect [Figure 5a-e] (P < 0.01). These findings suggested that mitochondrial autophagy plays a protective role in mitigating NLRP3-mediated cellular injury.

Export to PPT
JC-1 staining was performed to measure MMP in HUVECs to further assess the protective role of mitophagy in cellular injury [Figure 5f and g]. Compared with the siNC group, the siNLRP3 group exhibited increased red fluorescence, suggesting that NLRP3 silencing improved mitochondrial function. Following 3-MA treatment, red fluorescence decreased while green fluorescence increased. Meanwhile, Rapa enhanced red fluorescence, indicating that Rapa-induced mitophagy preserves the MMP. These findings indicated that mitophagy plays a pivotal role in NLRP3-mediated pyroptosis, and its activation preserves mitochondrial function while attenuating L-NAME-induced endothelial cell injury.
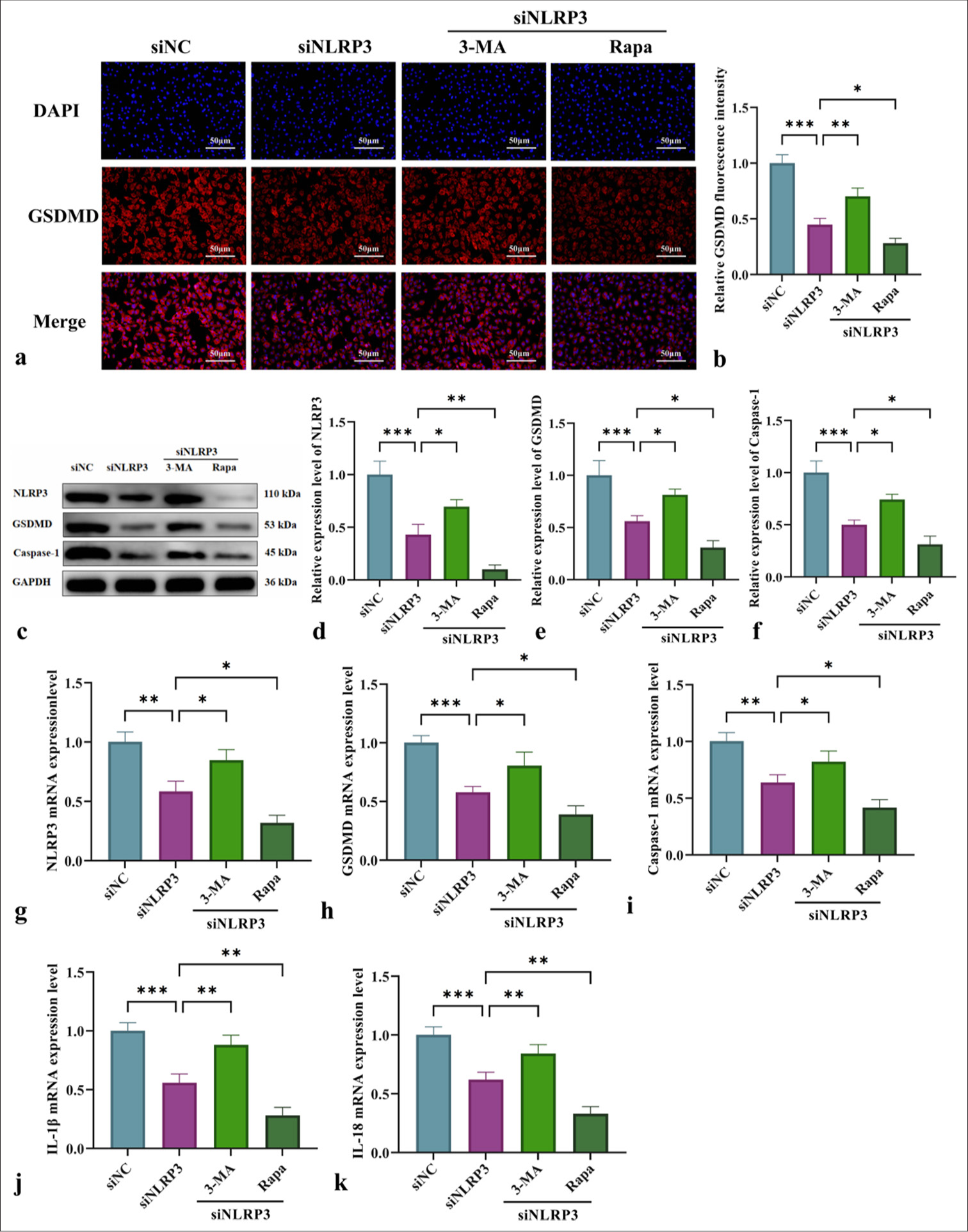
Mechanistic study of NLRP3 silencing combined with mitophagy regulation in pyroptosis and inflammatory cytokine releaseThis study examined the impact of NLRP3 silencing combined with the mitochondrial autophagy modulators 3-MA and Rapa on L-NAME-induced pyroptosis and inflammation in HUVECs. Immunofluorescence analysis [Figure 6a and b] demonstrated that NLRP3 silencing (siNLRP3 group) reduced GSDMD fluorescence intensity. Meanwhile, 3-MA enhanced GSDMD fluorescence intensity, and Rapa significantly suppressed it. WB and qRT-PCR analyses further confirmed that NLRP3 silencing (siNLRP3 group) downregulated NLRP3, GSDMD, and caspase-1 and reduced the release of pro-inflammatory cytokines IL-1β and IL-18 [Figure 6c-k]. Furthermore, the modulation of mitochondrial autophagy with 3-MA exacerbated pyroptosis and inflammation, but Rapa significantly mitigated these effects [Figure 6] (P < 0.05 or P < 0.01). These findings underscored the critical role of mitochondrial autophagy in regulating NLRP3-driven pyroptosis and inflammation.
Export to PPT
DISCUSSIONHypertensive disorders during pregnancy, including eclampsia, pre-eclampsia, and PIH, are common conditions and contribute significantly to maternal deaths on a global scale.[23] Gestational hypertension is often characterized by proteinuria, edema, and vascular endothelial damage. E
Comments (0)