
Remember me





Fibrosing mediastinitis (FM), also known as sclerosing mediastinitis or mediastinal fibrosis, is a rare condition characterized by local expansion of fibroinflammatory tissue within the mediastinum [1,2,3] (Fig. 1). Although there are no defined categories of fibrosing mediastinitis, it is usually divided into two sub entities – granulomatous and non-granulomatous; of the two sub-types, granulomatous has been reported to comprise of 80–90% of all fibrosing mediastinitis cases [2]. The condition tends to affect young patients with a slight female predilection. One of the leading causes of fibrosing mediastinitis is prior infection with Histoplasma capsulatum. Other infections, autoimmune conditions, prior radiation, and lymphoma are among some of the additional conditions associated with the development of FM. The symptoms associated with the condition vary considerably and largely depend on the organ involvement and extent of the fibrotic changes. Overall, there is still scant literature summarizing the etiologies and progression of fibrosing mediastinitis. In this report, we provide a review of the clinical manifestations, diagnostic evaluation, therapeutic modalities, and prognosis for patients with fibrosing mediastinitis.
Fig. 1
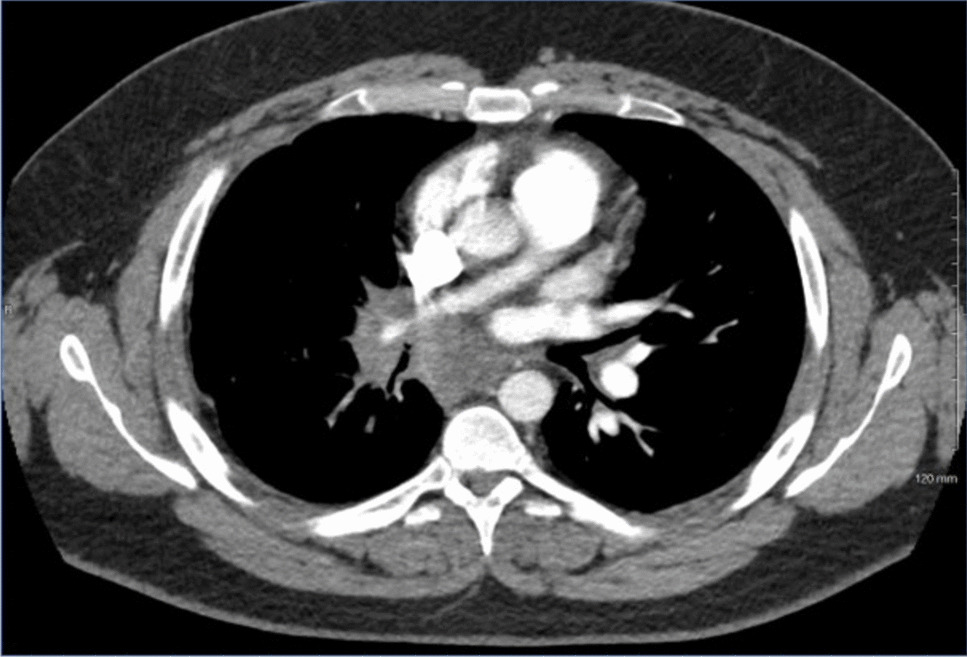
Fibrosing mediastinitis involving right hilum/mediastinum causing narrowing of the right pulmonary artery and bronchus intermedius
EpidemiologyTypically, patients who present with symptoms of fibrosing mediastinitis are young, with the average age range documented between 20–40 years of age [4, 5]. Initially, when the disease was first documented in literature in 1855, it was believed to be a rare process caused by syphilis or tuberculosis [3]. Since the mid-1900s, there have been more studies examining the etiology of fibrosing mediastinitis. In endemic areas, such as the mid-western region of the United States, Histoplasmosis is the overwhelming leading cause of fibrosing mediastinitis – comprising 78% of all cases in the United States (Fig. 2) [1,2,3, 6]. However, it should be noted that less than 1% of patients with a history of Histoplasmosis exposure are found to have developed fibrosing mediastinitis. Although usually thought of as a late complication of Histoplasmosis, one study estimated that in patients with Histoplasmosis, approximately 3 per 100,000 developed FM during the acute phase of the illness. Outside of Histoplasmosis, other infections that have been described to lead to the formation of fibrosing mediastinitis include Blastomycosis, Cryptococcus, and Aspergillosis [1,2,3, 6]. Other causes of fibrosing mediastinitis include idiopathic, autoimmune diseases, prior radiation therapy, Hodgkin lymphoma, rheumatic fever, trauma, drugs (methysergide, ergot drugs, bromocriptine, paclitaxel), IgG4-related disease, and Behcet’s disease [1,2,3, 6,7,8]. Of all noninfectious causes, idiopathic FM constitutes 58% of those cases.
Fig. 2
Etiologies of Fibrosing Mediastinitis. Histoplasmosis account for 78% of fibrosing mediastinitis cases followed by Idiopathic at approximately 11.6%. Less common etiologies include other infectious (~ 4%), autoimmune (~ 4%), and other (~ 2%)
Mechanisms/PathophysiologyInstead of thinking of fibrosing mediastinitis as an isolated disease, it should be considered a clinical pathologic syndrome driven by an underlying infectious or inflammatory cause. Fibrosis can occur in a variety of diseases, but the underlying mechanism in this condition is still poorly understood. Some studies have examined that the process may be in part caused by cytotoxic CD4 + T cells [9], which has been proposed to cause a delayed type IV hypersensitivity response [10]. Particularly in the setting of Histoplasmosis-associated FM, it has been proposed that cytotoxic CD4 + T cells recognize the H. capsulatum antigen leading to apoptosis within the mediastinum. In turn, this then contributes to excessive tissue remodeling with activation of macrophages and myofibroblasts [9].
Another study proposed that CD20 positive B lymphocytes accumulate in the mediastinum in response to an inciting factor (i.e. infection or inflammation) and its corresponding systemic depletion leads to an inflammatory cascade leading to fibrosis [10]. Activated B cells may also contribute to the reactivation of cytotoxic CD4 + T cells within mediastinal tissues [9].
Diagnosis, Screening, and PreventionClinical FeaturesThe abnormal fibro-proliferative response to an inflammatory stimulus leads to progression and eventual encasement of mediastinal structures (Fig. 3). The subsequent compression, encasement, and obstruction of vital mediastinal structures, such as the esophagus, airways, pulmonary arteries and veins, and central systemic veins leads to manifestations of symptoms [1, 5].
Fig. 3
Multifocal narrowing of the right central airways and post-obstructive atelectasis of the right middle lobe with persistent moderate to severe narrowing of the right main pulmonary artery
Common symptoms include cough, dyspnea, and pleuritic chest pain [1,2,3, 6,7,8,9,10]. When the airways are obstructed, it can lead to post-obstructive pneumonia. This progression can also lead to invasion of the pulmonary bronchus with fibrous tissue and pulmonary hypertension from compression of the pulmonary vasculature (Fig. 4). This obstruction of the arteries can lead to creation of anastomosis in the bronchial and intercostal arteries, which increases the risk of hemoptysis and occurs in approximately 20% of patients. The calcification that also occurs with the disease process can lead to broncholiths which pose a risk of eroding into the airways and blood vessels and causing massive hemoptysis as well [11].
Fig. 4
Chronic occlusion of the left inferior pulmonary vein and marked narrowing of the left pulmonary artery with fibrotic changes secondary to fibrosing mediastinitis through autoamputation
Rarer manifestations also include compression of the recurrent laryngeal nerve causing hoarseness of the voice [4]. The invasion in and around the pericardium can lead to constrictive pericarditis. Invasion around the larger systemic vessels can cause superior vena cava syndrome. Also, the presence of a pleural effusion may be suggestive of fibrosing mediastinitis. Typically, these effusions are unilateral, refractory, transudative effusions caused by systemic backflow of pressure [12]. If compression or invasion of the thoracic duct is involved, it can lead to the formation of a chylothorax [13].
Laboratory and Radiographic FindingsThere are no unified standards or criteria for diagnosis. Instead, there are varied clinical and histologic definitions used in various sources of literature [14]. Given that the most prevalent etiology of fibrosing mediastinitis is associated with infection, in particular Histoplasmosis, it is imperative to obtain a thorough travel and exposure history in order to ensure that prior infections have been ruled out [10]. Serologic workup including fungal serologies and IgG4 levels should also be checked in all individuals with findings that raise concerns for FM.
Radiographically, there are several findings that can help support the diagnosis of fibrosing mediastinitis. In a retrospective study of 33 patients, Park et al. established two distinct radiologic patterns—localized and diffuse patterns [15]. Among the patients studied, 82% demonstrated localized patterns with 63% of those having calcifications (Fig. 5). Typically, this localized pattern has been described in other case series and retrospective analysis with lymphadenopathy only visualized in the paratracheal and subcarinal region [11]. Conversely, the diffuse radiographic pattern is described as noncalcified lymphadenopathy impacting multiple mediastinal regions, with fibrosis spreading to other anatomical areas outside of the mediastinum [11].
Fig. 5
Large calcified left hilar and suprahilar nodal mass
Initial screening for Fibrosing Mediastinitis may include chest radiographs and/or CT scans of the chest. On chest radiographs, findings most prevalent with FM include widening of the mediastinum, atelectasis, mediastinal masses, hilar fullness, and pulmonary congestion [7]. On CT, examining for calcified mediastinal lesions or larger mediastinal lymphadenopathy should be performed with special note looking for compressions of mediastinal structures [2] (Fig. 6). Additionally, it should be noted that to adequately visualize the pulmonary vasculature, it is imperative to order a contrast enhanced CT of the chest with the appropriate protocol for better diagnostic accuracy. Notably, malignancies can mimic FM on CT and should also be considered in the appropriate clinical context [16]. Other associated imaging findings include volume loss, post-obstructive pneumonitis, and peripheral endoluminal mucus plugs [3, 4].
Fig. 6
Effacement of the right ventricle secondary to fibrosing mediastinitis
Additional imaging modalities have also been used to evaluate fibrosing mediastinitis. MRI is useful in patients who have iodinated contrast allergies or renal disease, but it is not specific for identifying calcifications [2, 13, 17]. FDG PET/CT is helpful in differentiating malignancy if there is no uptake, but can also show active inflammatory lesions, which has been associated with better disease response to treatment [2]. Similar to CT scan, caution should be used when using PET/CT as uptake on imaging could also raise concern for malignancy and misdiagnosis [17]. Angiography is typically not required for the diagnosis for FM.
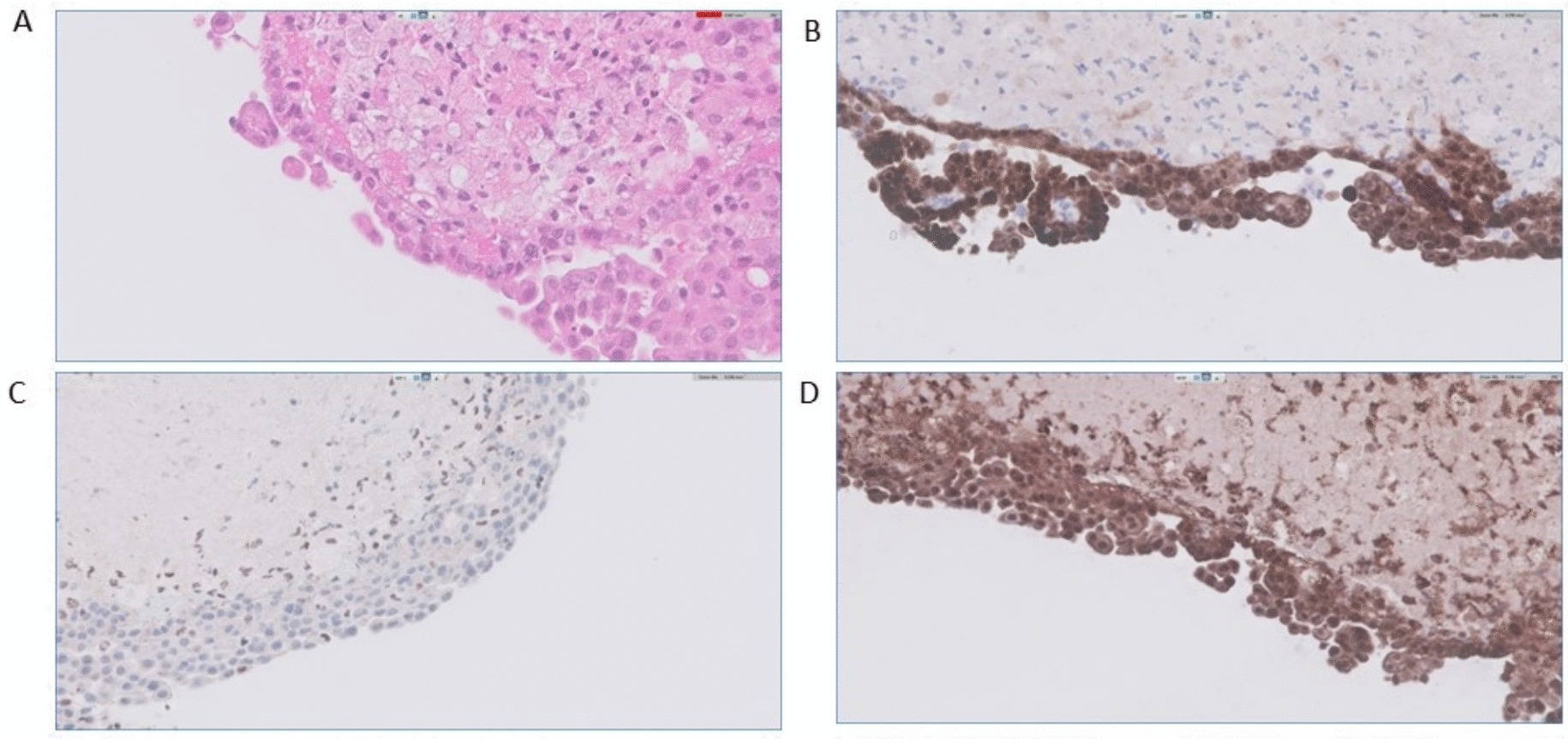
If the diagnosis remains unclear, biopsy has been recommended to confirm the diagnosis. EBUS is generally preferred as it is less invasive but can alternatively use mediastinoscopy/surgical exploration to obtain adequate tissue sampling [2, 17, 18]. When performing bronchoscopy, external compression of the airway can be noted with extensive erythema and mucus plugging (Fig. 7). Surgical biopsies obtained via thoracotomy have not only a high peri-operative mortality, with reports as high as 20%, but also may not have any significant benefit [19]. For example, in a series examining 44 patients who underwent thoracotomy, 9 total patients died, with 26 deemed unresectable, with another 12 requiring pneumonectomy, lobectomy, or airway reconstruction [19, 20]. Histopathologically, characteristics include abundant, paucicellular, fibrous tissue infiltrating and obstructing adipose tissue, with granulomas present in granulomatous fibrosing mediastinitis [1,2,3, 6]. Staging has also been proposed by Park et al. to consist of three stages: Stage I involving fibromyxoid tissue with numerous immune cells and lymphocytes, Stage II involving thick eosinophilic collagen with spindle cells and lymphocytes, and Stage III involving acellular collagen with lymphoid follicles and dystrophic calcification [15].
Fig. 7
Bronchoscopy with evidence of significant airway stenosis of the right main bronchus and bronchus intermedius secondary to large extrinsic compression by hilar/mediastinal adenopathy
When ordering and assessing imaging and diagnostic testing, caution should be used in interpreting the results given the variation of findings as well as the shared characteristics that fibrosing mediastinitis shares with other medical conditions. With Computed Tomography usually showing large mass-like mediastinal foci and PET/CT occasionally showing avid FDG uptake, patients are more likely to undergo more invasive testing and endobronchial biopsies to rule out malignancy, which then in turn further delays the diagnosis and treatment of fibrosing mediastinitis.
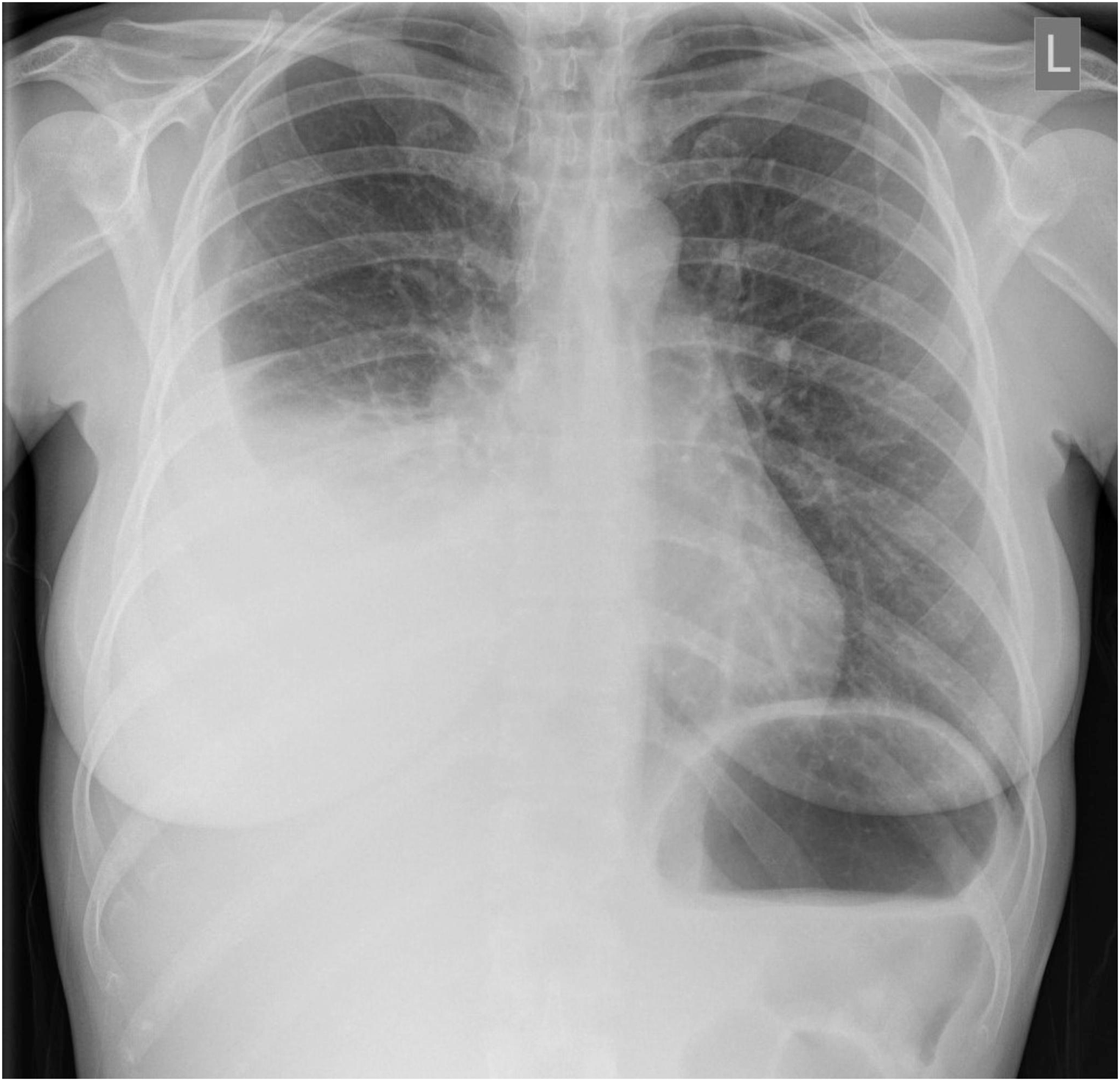
Pulmonary Hypertension in Fibrosing MediastinitisPulmonary hypertension caused by fibrosing mediastinitis is a pathologic condition caused by compression of the pulmonary arteries, veins, or both, which is characterized as a WHO group 5 condition [7] (Fig. 8). The elevated pulmonary pressures leading to right heart failure is a prevalent sequela leading to death among patients with FM. EKG and echocardiogram findings are non-specific but are imperative to screen patients for right axis deviation, right heart strain, right ventricular dilation, and reduced systolic function. Radiographically, looking for the FM Dyad (prominent main PA/enlarged RV plus atelectasis) or the FM Triad (FM Dyad plus refractory pleural effusion) have been suggestive of pulmonary hypertension. V/Q scan can show perfusion defects, which can be hard to differentiate from CTEPH; in these cases, SPECT/CT has been shown to be a better modality to differentiate between the two disease processes [7].
Fig. 8
Cardiac MRI showing chronic left pulmonary vein stenosis
When examining involvement of the pulmonary vessels, it has been characterized as three separate types: Type I involves stenosis in the pulmonary arteries, mostly accompanied by the stenosis of anatomically adjacent bronchi but not the pulmonary veins; Type II involves stenosis of the pulmonary veins without involvement of the pulmonary arteries, with bronchus involvement being rare; Type III involves stenosis of the pulmonary arteries, pulmonary veins, and bronchi [7].
Studies examining patients with fibrosing mediastinitis induced pulmonary hypertension have been sparse. The largest study was performed by Seifert et al., which was a retrospective observational study in a single center in France examining 27 patients from 2003–2014 [21]. The findings of the study found that 59% of patients had severe exercise limitations corresponding to NYHA class III and IV. Their Median six-minute walk test was 338 m. On right heart catheterization, the median mean pulmonary artery pressure was 42, with the values ranging from 27–90 mmHg, and a median PVR of 6.2 WU corresponding to severe disease [21].
Comments (0)