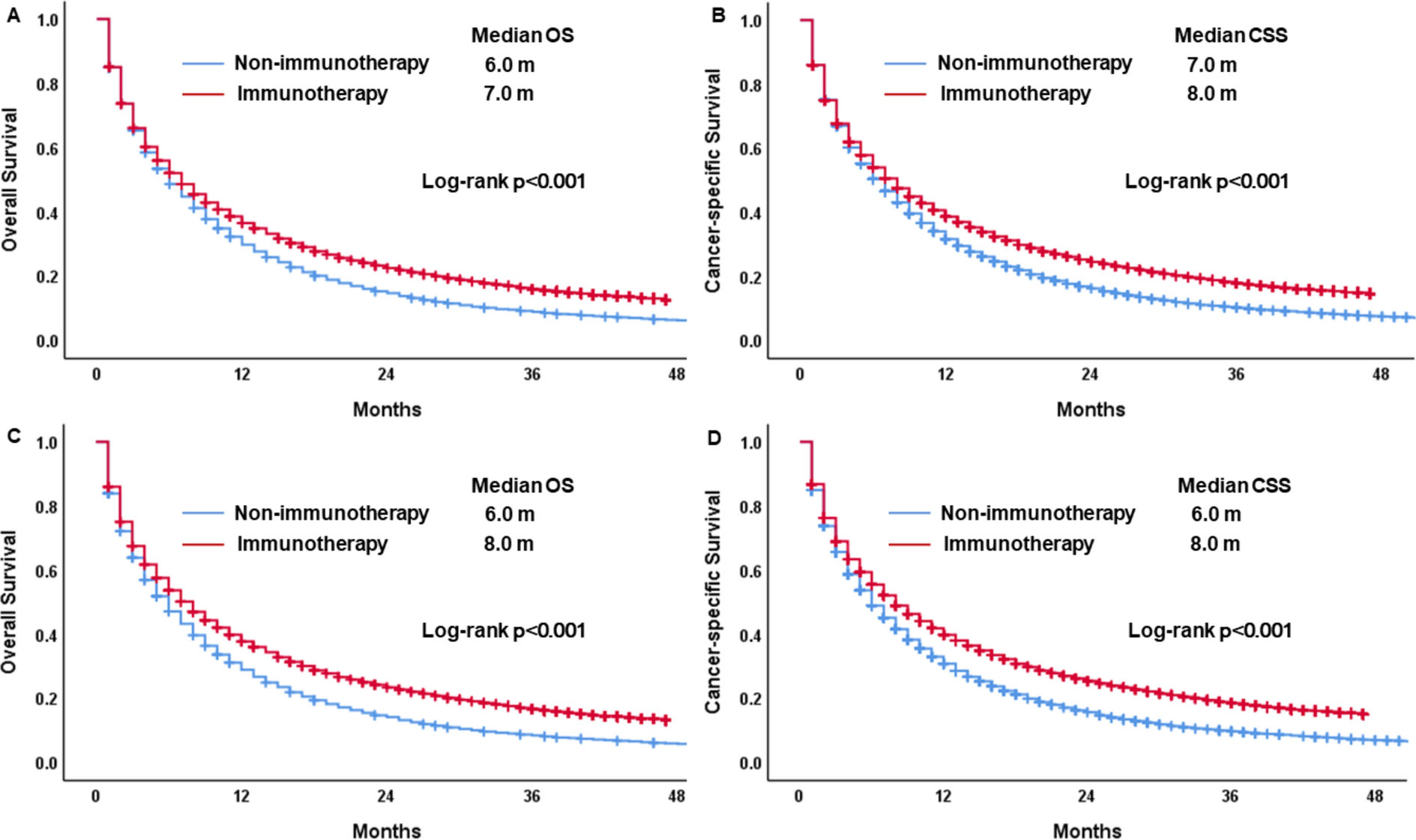
This was a multicenter, randomized, open-label, crossover study. Inclusion criteria for subjects required they be ≥ 65 years without severe renal impairment. Subjects were then stratified by no, mild, or moderate renal impairment. Renal function was defined by creatinine clearance. Those with no renal impairment had creatinine clearance > 90 mL/min. Creatinine clearance between 60 and 89 mL/min defined mild renal impairment, while creatinine clearance of 30–59 mL/min defined moderate renal impairment. Subjects with creatinine clearance < 30 mL/min were excluded.
This was a prospective study. Subjects were randomized to receive an intravenously administered bolus dosing regimen of ketorolac tromethamine (reference) or a loading dose/continuous infusion of intravenously administered NTM-001. Based on the approved ketorolac IV bolus dosing regimen of 15 mg every 6 h or 60 mg over 24 h, the continuous infusion group used a 6.25 mg loading dose of NTM-001 followed immediately by continuous infusion of 1.75 mg/h delivered using a preprogrammed conventional infusion pump. NTM-001 was administered as a continuous IV infusion over 24 h. Based on a model of ketorolac injection, an exposure–response modeling and simulation were employed to predict the time course of drug exposure and its analgesic effect, comparing NTM-001 to a reference regimen of IV bolus ketorolac tromethamine doses of 15 mg every 6 h over 24 h.
Subjects were stratified into three groups, those with no, mild, or moderate renal impairment. Each of these groups was studied in a randomized crossover design, allowing for a 1-week washout between doses. The first group (no renal impairment) was dosed sequentially followed by a parallel dosing plan for the renally compromised subjects.
Subjects were initially screened over a period of up to 28 days and then given a baseline assessment on day 1 when they entered the study. There were four treatment periods per subject and each treatment lasted 24 h. Blood sampling was done during the dosing and continued for 96 h after dosing. Between the four treatments, subjects had a washout period of at least 7 days.
Subjects were randomized to receive a half-dose of NTM-001 (6.25 mg loading dose, followed at once by 1.75 mg/h over 24 h continuous infusion) or a ketorolac tromethamine IV bolus (KETO-BOLUS) regimen of 15 mg delivered every 6 h over 24 h. The crossover design meant that subjects joined the opposite group after the initial dosing to act as their own controls.
The observed ketorolac concentration–time profiles were plotted on the model-predicted ketorolac target profile according to the PK base model. Concentration–time data from both NTM-001 and the KETO-BOLUS treatments were fitted with the base model using a nonlinear mixed-effects (NLME) model and Monolix 3.2 software (Lixoft, Atony, France). The median ± 95% population-predicted confidence intervals versus time were plotted over the corresponding target median and prediction intervals as set forth by the base model. The PK parameters were calculated from plasma concentration–time data using the noncompartmental methods with Phoenix® WinNonlin® version 8.1 software (Certara Company, Princeton, New Jersey, USA).
Adverse events were monitored and assessed for severity by the principal investigator. Predefined adverse events of special interest (AESIs) and stopping criteria were predefined. An independent data safety and monitoring board (DSMB) reviewed safety and exposure data on a continuous basis over the course of the study.
Protocols were approved by the IntegReview institutional review board in Austin, Texas. The study protocol is NTM-001-HP001. All patients signed informed consent before entry into the study. The study complied with the Good Clinical Practice requirements described in the current International Council for Harmonization (ICH) guidelines and current federal regulations. This study was not registered with ClinicalTrials.gov.
This study was performed in accordance with the Helsinki Declaration of 1964 and its later amendments.



Comments (0)