
Remember me



The institutional Review Board of the Ethical Committee approved this retrospective study (project number: 20–890, Goethe University Frankfurt am Main, Germany).
Study designOur study is an in-depth subgroup analysis of a previously reported patient cohort (Bernatz et al. 2020) with added novelty by epigenetic analysis, inclusion of a new control cohort with benign prostatic hyperplasia (BPH), and correlation with radiomics analysis. In short, 418 consecutive patients with confirmed PCa who had a mpMRI before radical prostatectomy (RPX) between 2014 and 2019 were screened for study inclusion to finally include a total of 30 patients (in comparison to the prior study (Bernatz et al. 2020) we had to exclude three patients with insufficient tissue quality for epigenetic analysis, therefore, resulting in 30 PCa patients). The further inclusion and exclusion criteria for the PCa patients are depicted in Bernatz et al. (2020). See Fig. 1 for the flow-chart of PCa-patient inclusion. Control patients were treated with holmium laser enucleation of the prostate (HoLEP) for BPH in 2019 and four patients were consecutively enrolled. The inclusion criteria for the control patients were (I) BPH, (II) no malignancy in pathologic analysis. Control exclusion criteria were (I) incidental malignancy in postoperative tissue specimens, (II) insufficient tissue quality. From four PCa patients, additional adjacent morphologically benign tissue was sampled for epigenetic analysis.
Fig. 1
STARD flowchart of prostate cancer patient inclusion into the study. The flowchart depicts the retrospective inclusion of the 30 prostate cancer patients as previously described (Bernatz et al. 2020). Four additional retrospective patients with BPH (median age 70 [61–76]) served as complete benign control patients which were consecutively enrolled in clinical routine
Reference standardAll tissue samples were histologically confirmed in the institution’s pathology department by a uropathologist (JK). All PCa and adjacent benign tissue samples were correlated with the matching localization in the mpMRI as previously described (Bernatz et al. 2020).
DNA methylation analysis and tumor deconvolutionThe tissue samples were subjected to DNA methylation analysis using the Human Methylation EPIC array by Illumina (Illumina, California, USA). Formalin-fixed, Paraffin-embedded tissue was cut in 4 μm thin section with a microtome (Leica SM 2000R, Wetzlar, Germany), mounted on slides (Superfrost Plus, Thermo Scientific, Braunschweig, Germany) and H&E stained. Representative sections of the lesions were selected, and punch biopsies (1.0 mm diameter, kai Europe GmbH, Solingen, Germany) were taken for DNA isolation by use of the Stratek Invisorb Genomic DNA Kit II (stratek molecular, Berlin, Germany). After assessment of DNA concentration using the Qubit DNA BR Assay Kit and Qubit 3 Fluorometer device (Invitrogen, Life Technologies Corporation, Oregon, USA), DNA was further processed and hybridized to the Human Methylation EPIC array beadchips (Illumina, California, USA) following standard protocols provided by the manufacturer. EPIC array beadchips were scanned by an iScan (Illumina, California, USA) and raw intensity data (idats) was obtained. Idats were imported into the R software package “RnBeads” (Müller et al. 2019) to perform quality control, exploratory and differential methylation analysis as well as to obtain LUMP estimates. The LUMP algorithm uses measurements of leucocyte unmethylation to infer leukocyte infiltration in bulk tissue samples by the analysis of 44 CpG sites which are unmethylated in leukocytes and methylated in tumor cells (Aran et al. 2015). DNA methylation data was normalized using the “dasen” method from the R package “watermelon”.
Reference-free deconvolution of prostate tissue was performed using MeDeCom, which uses non-negative matrix factorization to compute Latent Methylation Components (LMCs; Scherer et al. 2020). LMCs represent methylation patterns shared between the samples’ most variable CpG sites - i.e. the top 5000 most variable CpG sites across all samples of this study - with correction for methylation patterns driven by patient age. LMCs are selected by evaluating cross-validation errors for LMCs numbers (kappa) and the regularization parameter (lambda). For each sample, proportions of LMCs were computed and subjected to hierarchical cluster analysis by use of Ward’s minimum variance method. LMCs-based clusters were further correlated with clinical tumor parameters and their cellular composition.
For reference-based deconvolution of prostate tissue we used MethylCIBERSORT as described in (Chakravarthy et al. 2018). In brief, idats are loaded into R, assessed for quality, Noob normalized and beta value calculated by use of the minfi package. An in silico cellular mixture matrix is generated by combining signature CpGs of immune cells (T regulatory cells, CD4 + effector cells, CD8 + T cells, CD20 + B cells, CD14 positive monocytes, eosinophils, neutrophils, NK cells), fibroblasts, endothelia and cancer cells with the samples’ CpGs to infer the estimates of cellular fractions present in the prostate tissue. Deconvolution of the files was realized on the CIBERSORT X platform provided by the Alizadeh and Newman labs (Newman et al. 2015).
MRI imaging and examinationAll imaging was performed on a single 3-T scanner and read in clinical routine as previously described (Bernatz et al. 2020), following the European Society of Urogenital Radiology (ESUR) guidelines. For the radiomics analysis, the MR images (T2-weighted (T2w), apparent diffusion coefficient (ADC), dynamic contrast-enhanced (DCE) were exported in “Digital Imaging and Communications in Medicine” (DICOM) format. Representative images of mpMRI acquisition are depicted in (Bernatz et al. 2020) and acquisition parameters are depicted in Supplementary Table 1.
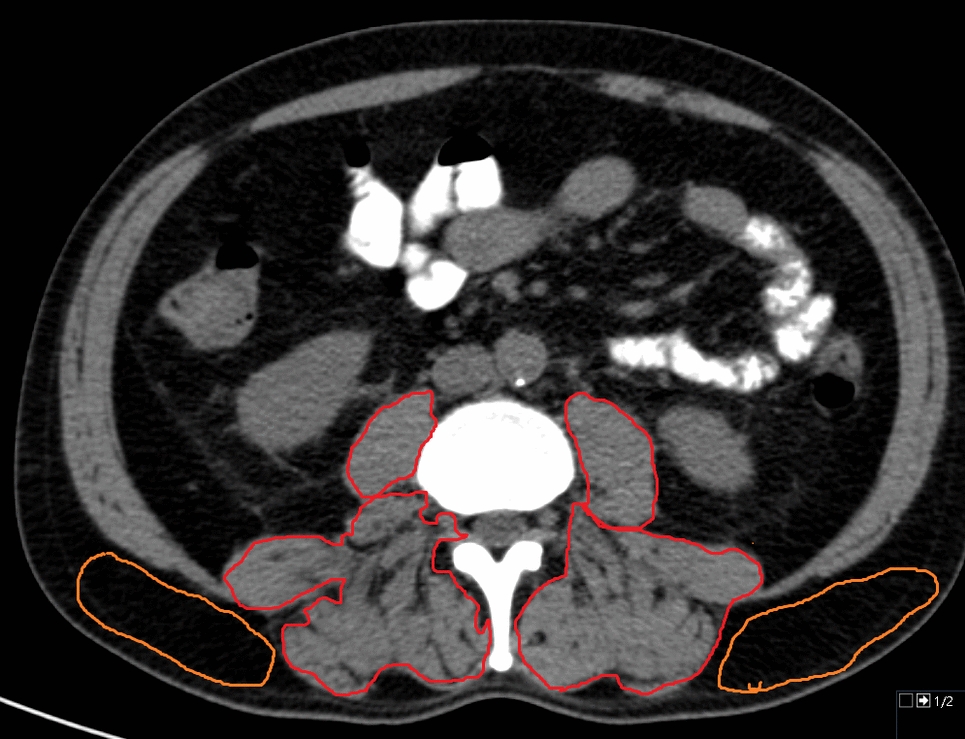
MRI segmentationWe depict the workflow of MRI segmentation in detail elsewhere (Bernatz et al. 2020). In short, we used the open-source 3D slicer computing platform (http://slicer.org, version 4.9.0) (Fedorov et al. 2012; Velazquez et al. 2013) to visualize and segment the whole 3-dimensional tumor volume of interest (VOI) of each tumor index lesion using ADC maps. Manual seeds were defined in each PCa index lesion with semi-automatic 3D-VOI annotation by grow-from-seeds algorithm (Velazquez et al. 2013; van Griethuysen et al. 2017). The benign adjacent tissue was manually defined. We depict representative images of the whole habitat index PCa lesion segmentation in Supplementary Fig. 1.
Feature extractionWithin the 3D Slicer software platform, we used the open-source extension PyRadiomics (Pedregosa et al. 2011; Velazquez et al. 2013) to extract 105 radiomics features of seven feature classes as previously described (Bernatz et al. 2020).
Quantitative radiographic biomarkers to predict epigenetic signaturesThe analysis included 30 PCa patients with matching pathologic and radiologic index lesions. The control (BPH) patients did not have a mpMRI and were excluded from the radiomics machine learning analysis. All analyses were performed in Python 3.9.16. We used Pearson correlation analysis to drop all highly correlated (r > 0.95) features (n = 70) to reduce the risk of overfitting and to stratify our final radiomic features set. We split our dataset into an independent training (70%) and testing set (30%) with patient samples drawn at random. We scaled the features using StandardScaler (Bernatz et al. 2023) to have a mean value of 0 and a variance of ± 1. Next, we independently applied a pool of four variant machine learning models to predict the epigenetic signature clusters. We used different established machine learning models (I) logistic regression (LR), (II) random forest (RF), (III) ada boost (ADB) and (IV) stochastic gradient boosting (SGB). The machine learning pipeline is described in detail elsewhere (Virtanen et al. 2020). For each model, we depict the receiver operating characteristics (ROC) area under the curve (AUC) as implemented in scikit-learn 1.0.2 (Pedregosa et al. 2011).
General statistical analysisStatistical analyses were performed in JMP (JMP Statistical Software, SAS Institute, Cary, North Carolina, USA), R (R Core Team 2021), and Python, using SciPy (SciPy.stats) (Virtanen et al. 2020) and scikit-learn (Pedregosa et al. 2011) for further statistical analyses. Graphical illustrations were performed in Affinity Designer 2.1 (Serif (Europe) Ltd). The PCa sample size resulted from including all eligible patients according to the inclusion and exclusion criteria (Bernatz et al. 2020).
Comments (0)