
Remember me





The Golgi apparatus was first reported as an “internal reticular apparatus” occupying the perinuclear region by Camillo Golgi in 1898. Its existence was later confirmed by Dalton and Felix using electron microscopy [1]. The Golgi apparatus is a highly polarized organelle [2]. Newly synthesized products from the endoplasmic reticulum (ER) enter the stacks from the cis-side of the Golgi and sequentially pass through various cisternae containing specific enzymes, undergoing post-translational modifications including glycosylation, acetylation, sulphation, phosphorylation, methylation, palmitoylation, and proteolytic cleavage [3], and finally arrive at the trans-Golgi network (TGN), where they are sorted into different vesicles and delivered to specific parts of the cell or secreted outside of it [4]. The classical functions of the Golgi apparatus, membrane transport and glycosylation, are carried out in separate stacks. In vertebrates, however, Golgi stacks collect near the minus end of microtubules, which are aligned by tubular structures and connected laterally to form Golgi ribbon [5]. This process relies on the participation of Golgi matrix proteins and the maintenance of intact microtubule organization [6]. The ribbon structure of the Golgi apparatus allows it to perform a variety of sophisticated functions. For instance, it allows Golgi glycosyltransferases to move laterally between adjacent stacked pools, ensuring precise protein glycosylation [7]. The Golgi ribbon also plays a crucial role in regulating mitotic progression [8], establishing and maintaining cellular polarization, and facilitating directional cell migration [9, 10], among other functions. Additionally, the Golgi is considered to be a central hub for various signaling pathways that regulate cellular processes. It is involved in several biochemical processes including DNA repair [11], stress response [12], control of ionic and reactive oxygen species (ROS) homeostasis [13], apoptosis [14], pro-inflammatory responses [15], and autophagy [16].
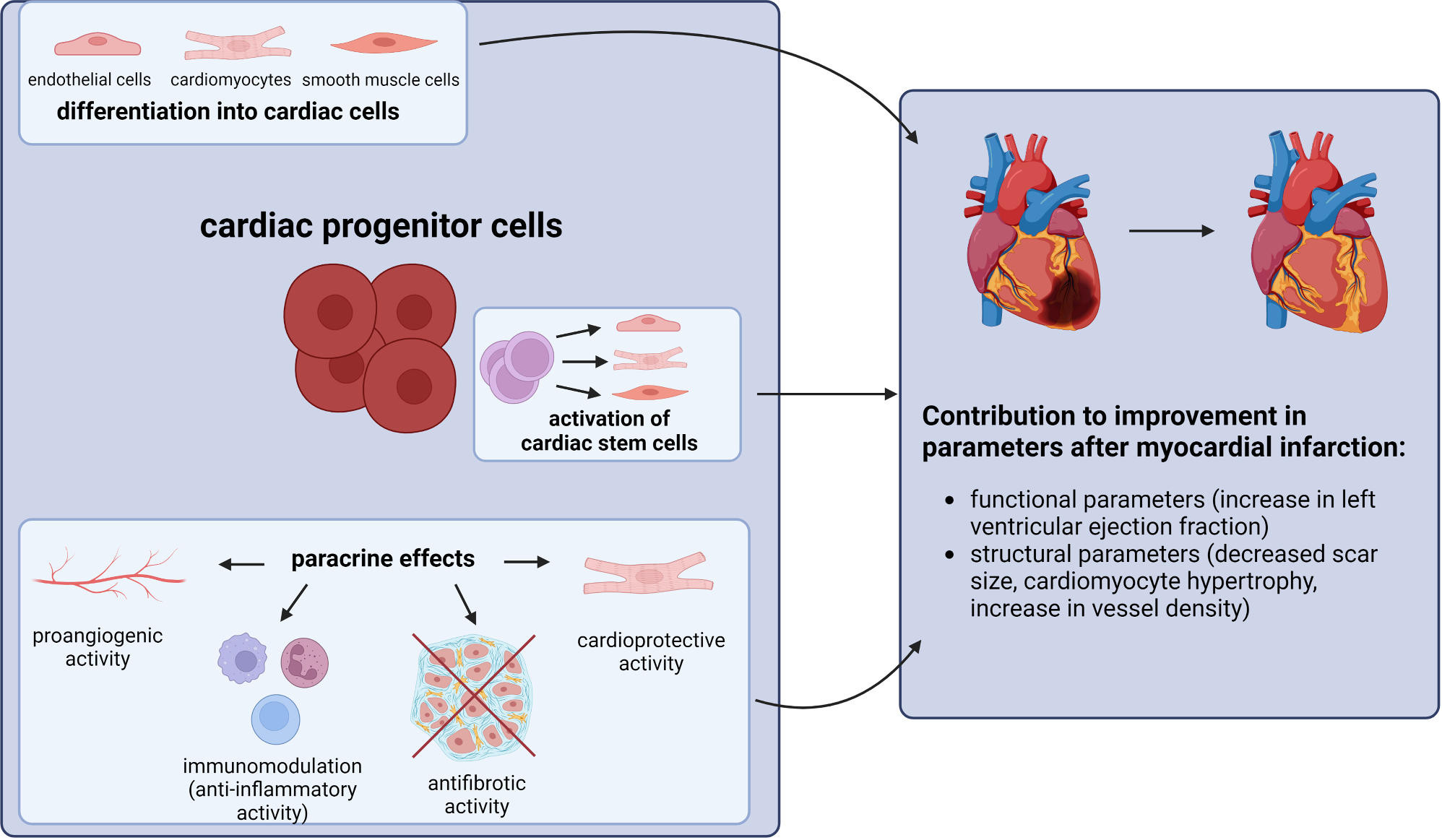
The Golgi apparatus is a highly dynamic organelle that is susceptible to fragmentation as a result of various pathological conditions. As early as 1966, disorganized Golgi structures were first identified in myeloma cells [17]. In the following decades, Golgi fragmentation was progressively observed in pathological conditions such as drug stimulation [18], viral infection [19,20,21], neurodegenerative diseases [22,23,24], and cancer [25,26,27]. As research advanced, a potential association between Golgi fragmentation and autophagy was identified. In 2011, Takahashi et al. found that under starvation stress, Bax-interacting factor 1 (Bif-1/Endophillin B1) regulates the transport of Atg9 vesicles from the Golgi apparatus to autophagosomes by mediating Golgi fragmentation and thus promotes autophagosome biogenesis [28]. Subsequently, Gosavi et al. demonstrated that the intact ribbon structure of the Golgi apparatus is the site of mammalian target of rapamycin (mTOR) localization and activation. The researchers discovered that the overexpression of the membrane tether coiled-coil domain containing 88 kDa (GCC88) across the Golgi network resulted in the rupture of the Golgi ribbon, which in turn led to the inhibition of mTOR and a subsequent increase in autophagy levels [29]. Initially, autophagy was thought to maintain cellular homeostasis by non-selectively degrading cytoplasmic components in response to stress. However, more and more studies have demonstrated that cells are capable of exclusively or preferentially degrading specific damaged organelles through selective autophagy. It was the year 2020 when Lu et al. demonstrated that fragmented Golgi is found to accumulate around and be engulfed by autophagosomes during nutrient starvation, thus the concept of Golgiphagy was proposed to describe the process of depletion of fragmented Golgi or Golgi components through selective autophagy [30]. Since then, Golgiphagy has gradually attracted the attention of researchers, and different proteins have been identified to function as Golgiphagy receptors. A new targeted drug against Golgiphagy has also been developed recently (Fig. 1). However, the specific mechanism of Golgiphagy seems to be still being explored.
Fig. 1
The key discoveries in the Golgiphagy field
Core machinery of autophagyAutophagy is a highly conserved catabolic process that depends on the lysosomal pathway during the long-term evolution of eukaryotic cells [31]. According to different mechanisms and functions, autophagy is divided into three forms: macroautophagy, microautophagy, and chaperon-mediated autophagy (CMA). Microautophagy is a process whereby specific cytoplasmic components and other intracellular components are directly engulfed by lysosomes for subsequent degradation [32]. CMA is a process by which a cytoplasmic chaperone protein, the heat shock cognate 71-kDa protein (HSC70), binds to a cytoplasmic protein containing a KFERQ or KFERQ-like motif in its amino acid sequence, and then brings this substrate protein to the surface of the lysosome for internalization and rapid lysosomal degradation [33]. Macroautophagy (hereinafter referred to as autophagy) is different from them in the formation of autophagosomes. The key to autophagosome formation in mammals is the initiation of the UNC51-like kinase 1 (ULK1) complex.
In conditions of nutrient starvation, hypoxia, and oxidative stress, the ULK1 complex is activated due to the inactivation of mTOR and the activation of Adenosine 5’-monophosphate-activated protein kinase (AMPK), as well as autophosphorylation of the ULK1 complex. The formation of autophagosomes is facilitated by the recruitment of multiple copies of activated ULK complexes to the sites of autophagosome formation on the ER [34]. The recruitment of ULK1 complexes may involve multiple mechanisms. For example, Vesicle Associated Membrane Protein-associated protein A/B (VAPA/VAPB) could recruit ULK1 complexes through direct interaction with FIP200. GABARAP also binds ULK1 and thus activates and recruits ULK1 complexes to promote autophagy initiation [35].
Additionally, autophagosomes are formed by the contribution of ATG9-containing vesicles derived from TGN. ATG9A is subject to regulation by AMPK- and ULK1-mediated phosphorylation under different conditions, which in turn influences the extent of autophagy [36]. ATG9A can also be recruited to autophagosome forming sites through interactions with ATG13-ATG101, which are part of the ULK1 complex [37].
Subsequently, the class III phosphatidylinositol-3-kinase (PI3KC3) complex I targets the autophagosome formation site in the presence of the ULK1 complex, thereby generating phosphatidylinositol 3-phosphate (PI3P). PI3P can recruit the β-propellers that bind polyphosphoinositides (PROPPIN/WIPI) family, of which WIPI2 is of particular functional importance. The ATG12-ATG5-ATG16L1 complex is recruited to the phagophore membrane by WIPI2B and WIPI2D or by interaction with FIP200, and it can exert E3 enzyme activity to promote the lipidation of mammalian ATG8 family proteins. The ATG8 family proteins may drive phagophore membrane expansion in different ways and play a crucial role in phagophore closure, as well as the subsequent fusion of autophagosomes and lysosomes [34, 35, 38].
Notably, WIPI3 or WIPI4 may interact with ATG2 and recruit it to the phagophore membrane, contributing to lipid transport between the ER and phagophore and promoting phagophore expansion. Meanwhile, ATG2A also interacts with ATG9, phospholipid recombinase vacuole membrane protein 1 (VMP1) and transmembrane protein 41B (TMEM41B) on the ER to form a lipid transfer unit, which maintains equilibrium in the density of phospholipids on each leaflet during lipid transfer [39].
Then, the autophagosome is closed by the action of the endosomal sorting complex required for transport (ESCRT) complex [40, 41]. Subsequently, fusion of autophagosomes and lysosomes occurs mainly in the presence of soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) proteins, homotypic fusion and vacuole protein sorting (HOPS) complexes, and small GTPases such as RAB7. Following fusion, the cargo is degraded by hydrolytic enzymes within the lysosome, and the degradation products are then reused by the cell [39, 42] (Fig. 2).
Fig. 2
Core machinery of autophagy. In response to various stress conditions, the mTORC1 and AMPK pathways regulate the kinase activity of the ULK1 complex, thereby initiating autophagy. The activated ULK1 complex is recruited near the ER membrane due to the interaction of FIP200 with VAPA/VAPB on the ER. Additionally, ATG9 vesicles are recruited to the ER membrane to provide a membrane source through interaction with the ATG13-ATG101 subcomplex. Subsequently, ULK1 further activates the PI3KC3 complex 1, resulting in the generation of PI3P. WIPI2 then binds to PI3P and further recruits the ATG12-ATG5-ATG16L1 complex to phagophore, thereby mediating ATG8 lipidation. Concurrently, PI3P also recruits WIPI3 or WIPI4 to phagophore, where WIPI3/4 transfer phospholipids from the ER to phagophore through interactions with ATG2, ATG9, as well as VMP1 and TMEM41B on the ER, thereby promoting phagophore expansion. Subsequently, the autophagosome closes through the action of the ESCRT mechanism, after which specific SNARE proteins, HOPS complexes, and small GTPases such as RAB7 mediate the fusion of the autophagosome with the lysosome. Following fusion, the cargo is degraded by hydrolytic enzymes within the lysosome and reused by the cell. Abbreviations: mTORC1, mammalian target of rapamycin complex 1; AMPK, adenosine 5’-monophosphate-activated protein kinase; ULK1, UNC51-like kinase; ER, endoplasmic reticulum; FIP200, focal adhesion kinase family interacting protein of 200 kD; VAPA, VAMP (Vesicle Associated Membrane Protein)-associated protein A; VAPB, VAMP (Vesicle Associated Membrane Protein)-associated protein B; ATG, autophagy-related; PI3KC3, phosphoinositide 3-kinases catalytic subunit type 3; PI3P, phosphatidylinositol 3-phosphate; WIPI, WD repeat structural domain phosphatidylinositol-interacting protein; VMP1, Vacuole membrane protein 1; TMEM41B, Transmembrane Protein 41B; ESCRT, endosomal sorting complex required for transport; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptors; HOPS, homotypic fusion and vacuole protein sorting; RAB, Ras analog in brain
Involvement of the golgi apparatus in autophagyThe Golgi, as a central intracellular transport hub, also plays a crucial role in autophagy. Firstly, the Golgi could act as a contributor to the autophagosome membrane to promote autophagosome biogenesis [43, 44]. This may be attributed to the formation and translocation of ATG9 vesicles (Fig. 3). ATG9, the sole multi-transmembrane protein in the ATG family of proteins, is predominantly resides in the membranes of the TGN and endosomes. Geng et al. demonstrated that after autophagy induction two post-Golgi proteins, Sec2 and Sec4, could direct the flow of Golgi-derived Atg9 vesicles to the autophagosome formation site [45]. Also, Yamamoto et al. demonstrated that Golgi-derived Atg9 vesicles were integrated into the outer membrane of autophagosomes [46]. Moreover, the transport of ATG9 vesicles from the Golgi apparatus to the autophagosomes can be regulated by different protein components. For instance, Bif-1, which was previously mentioned, can regulate starvation-induced Golgi membrane fission and the redistribution of Atg9 to the peripheral cytoplasm, which also requires the activity of PI3KC3 [28]. Additionally, ATG9A vesicles from the Golgi can transport active phosphatidylinositol-4-kinase-IIIβ (PI4KIIIβ) to the autophagosome initiation site under the regulation of Arfaptin2, which regulates the production of phosphatidylinositol 4-phosphate (PI4P) on the phagosome membrane and thus promotes autophagy [47]. In response to starvation stress, ULK1 phosphorylates ATG9A to promote the interaction of ATG9A with the adaptor protein 1 (AP1) complex, which in turn promotes the movement of ATG9A from the TGN to the ATG9A compartment and facilitates the initiation of autophagy [48]. Whereas, when ATG9A export from the TGN is impeded due to AP4 deletion, the delivery of ATG9A vesicles to autophagosomes is diminished, thereby impairing autophagosome formation [49, 50].
Fig. 3
The involvement of Golgi in ATG9 trafficking during autophagy. ATG9 vesicles are transported from the Golgi compartment to autophagosome formation sites (phagophore) via AP1 and/or AP4 complexes, which also deliver PI4KIIIβ. Furthermore, ULK1 phosphorylates ATG9, thereby facilitating ATG9 binding to AP1 complexes. The correct delivery of ATG9 vesicles to phagophore is also regulated by Arfaptin2, Bif-1, and the PI3KC3 complex. Abbreviations: ATG, autophagy-related; AP, adaptor protein; PI4KIIIβ, phosphatidylinositol-4-kinase-IIIβ; ULK1, UNC51-like kinase; Bif-1, BAX-interacting protein 1; PI3KC3, phosphoinositide 3-kinases catalytic subunit type 3
In addition, certain Golgi-related proteins are involved in regulating the process of autophagy. For example, the Golgi Re Assembly Stacking Protein 55 (GRASP55) de-O-GlcNAcylates upon glucose starvation and targets the autophagosome-lysosome interface through interactions with LC3-II and lysosomal associated membrane protein 2 (LAMP2), where it serves as a bridge to promote autophagosome-lysosome fusion [51]. GRASP55 also further promotes autophagosome maturation by facilitating the assembly of the PI3K UVRAG complex [52]. In response to autophagic stimuli, RAB2 dissociates from the Golgi and promotes phagophore formation by recruiting and activating the ULK1 complex. Subsequently, RAB2 shifts to interact with rubicon like autophagy enhancer (RUBCNL) and Syntaxin 17 (STX17) to further recruit HOPS complexes into autophagosomes to promote fusion with lysosomes [53]. Furthermore, Coat Protein Complex I (COP I) vesicles, which are involved in retrograde transport of proteins from the Golgi to the ER, have also been shown to play a role in autophagy. It has been shown that deletion of the COP I subunit leads to disruption of Golgi structure and accumulation of autolysosome-like structures in plant cells, which inhibit autophagy [54]. A recent study demonstrated that COP I vesicles function upstream of mTORC1 and activate autophagy by regulating the phosphorylation of S6 Kinase 1 (S6K1), which in turn plays a key role in the formation of autophagosomes during mineralization [55].
Golgiphagy receptorsThe Golgi apparatus is not only involved in the regulation of autophagy, but can itself be selectively degraded as an autophagic cargo. In recent years, research has identified several proteins that may act as Golgiphagy receptors to mediate the degradation of Golgi components through the autophagic pathway. These include Golgi phosphoprotein 3 (GOLPH3), calcium binding and coiled-coil domain protein 1 (CALCOCO1), Golgi microtubule-associated protein (GMAP), as well as members 3 and 4 of the Yip1 domain family (YIPF3 and YIPF4). The following provides a detailed description of the Golgiphagy receptors that have been studied in recent years.
GOLPH3GOLPH3 is also referred to GMx33, GPP34, or MIDAS, and its yeast homologue is Vps74p. GOLPH3 is a highly conserved protein initially identified in a proteomic characterization of the rat Golgi apparatus. It is subsequently recognized as a Golgi matrix protein, which is mainly enriched at the trans-side of the Golgi apparatus through the conserved C-terminal domain dubbed GPP34 [56, 57](Fig. 4A). GOLPH3 has multiple functional roles in cells. GOLPH3 can be rapidly exchanged between the cytoplasmic and Golgi-associated pools. Additionally, it has been found to be associated with tubules and vesicles that leave the Golgi apparatus [58]. Dippold et al. discovered that GOLPH3 is crucial in anterograde trafficking from the Golgi apparatus to the plasma membrane. Furthermore, GOLPH3 can specifically attach to the Golgi membrane by binding to PI4P and myosin XVIIIA (MYO18A), thus maintaining the flat appearance of the trans-Golgi [59]. GOLPH3 also maintains the localization of specific glycosyltransferases within the Golgi apparatus [60,61,62].
Fig. 4
The structure of the Golgiphagy receptors. (A) Schematic structure of GOLPH3 protein. GPP34, PI4P-binding domain. (B) Schematic structure of CALCOCO1 protein. SKICH, SKIP carboxyl homology domain; LIR, LC3-interacting region; CC, coil-coil region; zDABM, zDHHC-AR-binding motif; UIR, UDS-interacting region; ZF, Zinc Finger; FFAT, two phenylalanines in an acidic tract domain. (C) Schematic structure of dGMAP protein. GRAB, GRIP-related Arf-binding domain. (D) Schematic structures of the YIPF3 protein and the YIPF4 protein. TMD, transmembrane domain. (E) A table of the amino acid sequences as well as the positions of the LIR motifs in these Golgiphagy receptors
Lu et al. demonstrated that GOLPH3 may act as a cargo receptor to target the Golgi apparatus to autophagosomes for lysosomal degradation [30] (Fig. 5A). First, their study revealed that under conditions of starvation, treatment with various Golgi stress inducers, such as Brefeldin A, Nocodazole, Monensin, and Nigericin, resulted in increased co-localization of GM130-RFP (a cis-Golgi marker) and TGN46-RFP (a trans-Golgi marker) with LC3B-GFP (an autophagosome marker). Furthermore, transmission electron microscopy revealed that starvation treatment results in the accumulation of Golgi fragments around autophagosomes and phagocytosis by autophagosomes, supporting the occurrence of Golgiphagy and suggesting that Golgi stress inducers may promote Golgiphagy. Additionally, it was discovered that endogenous GOLPH3 in H9c2 cells, HUVECs, and HA-VSMCs cells can interact with LC3B under normal, starvation, or hypoxia-stimulated conditions. Moreover, the knockdown of GOLPH3 resulted in a decrease in the co-localization of Golgi marker proteins with LC3B in H9c2 cells, HUVECs, and HA-VSMCs. This led to the speculation that GOLPH3 may function as a Golgiphagy receptor. However, further investigation is required to elucidate the molecular mechanism underlying GOLPH3’s function as a Golgiphagy receptor.
Fig. 5
Models of Golgiphagy receptors-mediated Golgiphagy upon nutrient starvation in mammalian or Drosophila melanogaster. (A) GOLPH3, which is located on the trans-Golgi by binding PI4P, interacts with ATG8 to promote the encapsulation of Golgi fragments by phagophore. (B) CALCOCO1 localizes to the Golgi apparatus through its zDABM motif binding to the AR domain of ZDHHC17. Subsequently, CALCOCO1 interacts with ATG8 through its LIR and UIR motifs, recruiting the autophagy machinery and thus facilitating the degradation of Golgi fragments. (C) In Drosophila melanogaster, dGMAP can directly bind to ATG8 through the N-terminal LIR motif, thereby mediating the autophagy pathway degradation of the Golgi fragments. (D) YIPF3 and YIPF4, which are anchored to the Golgi through 5 tightly stacked transmembrane domains, form a heterodimer. As the only membrane-embedded Golgiphagy receptors, they interact with ATG8 to mediate Golgiphagy. Abbreviations: GOLPH3, Golgi phosphoprotein 3; CALCOCO1, calcium binding and coiled-coil domain protein 1; dGMAP, Golgi microtubule-associated protein in Drosophila melanogaster; YIPF3, the member 3 of Yip1 domain family; YIPF4, the member 4 of Yip1 domain family; PI4P, phosphatidylinositol 4-phosphate; AR, ankyrin repeat; SKICH, SKIP carboxyl homology; LIR, LC3-interacting region; CC, coil‐coil regions; zDABM, zDHHC-AR-binding motif; UIR, UDS‐interacting region; ZF, Zinc Finger; FFAT, two phenylalanines in an acidic tract
CALCOCO1CALCOCO1 is a paralogous homologue of two previously described autophagy receptor proteins, CALCOCO2/NDP52 and CALCOCO3/TAX1BP1. These three proteins constitute the CALCOCO family, sharing the same conserved domains: an N-terminal SKIP carboxyl homology (SKICH) domain, middle coil-coil regions (CC), and an atypical LC3-interacting region (LIR) motif [63]. Furthermore, CALCOCO1 contains a UDS-interacting region (UIR) that functions in conjunction with the LIR to bind LC3 [64](Fig. 4B). Previous research has demonstrated that CALCOCO1 is involved in transcriptional co-activation, glucose metabolism, and calcium signaling. Stefely et al. later expanded on the role of CALCOCO1 in mTOR-regulated selective autophagy, discovering through mass spectrometry proteomic analysis that CALCOCO1 may be a novel autophagy-associated protein. The authors also demonstrated that CALCOCO1 can physically interact with LC3. In addition, the deletion of the CALCOCO1 gene has been shown to disrupt ER-phagy [65]. Nthiga et al. demonstrated that CALCOCO1 is capable of mediating ER-phagy. CALCOCO1 interacts with VAPA and VAPB proteins in the ER through a novel FFAT-like motif and with the ATG8 family proteins through LIR and UIR motifs. This interaction recruits autophagic machinery to degrade cargo [64].
Recently, Nthiga et al. discovered that CALCOCO1 also regulates Golgi size and morphology by mediating Golgiphagy in eukaryotic cells through its interaction with the zinc finger DHHC-type palmitoyltransferase 17 (ZDHHC17) [66] (Fig. 5B). Under basal conditions, CALCOCO1 can interact with ZDHHC17 and ZDHHC13 located on the Golgi apparatus and thus anchored to it. This interaction is mediated by the zDHHC-AR-binding motif (zDABM) on CALCOCO1 with the ZDHHC17 N-terminal ankyrin repeat (AR) domains. Studies show that CALCOCO1 can recruit most of the ZDHHC17-containing Golgi fragments produced by starvation induction into autophagosomes and deliver them to lysosomes for degradation by interacting with LC3/GABARAP proteins. The authors demonstrated that the absence of interaction between CALCOCO1 and ZDHHC17, or the presence of a mutant CALCOCO1 lacking LIR and UIR motifs, leads to a reduction or impairment in the autophagic degradation of Golgi components. It is noteworthy that CALCOCO1-mediated Golgiphagy is induced by the need to remove excess Golgi components produced during stress in order to restore the pre-stress state of the Golgi apparatus. Under nutrient-sufficient conditions, constitutive Golgi turnover may not necessitate CALCOCO1-ZDHHC17-dependent degradation. Alternatively, it was discovered that TAX1BP1, which shares significant sequence similarity and identity with CALCOCO1, could also mediate its interaction with ZDHHC17 through the AR-zDABM interface. Therefore, it is worth investigating whether TAX1BP1 also functions in a similar mode in Golgiphagy.
GMAPHuman GMAP-210 (Golgi microtubule-associated protein 210) is a 210 kDa peripheral Golgi protein located in the cis-Golgi network (CGN), classified as a member of the golgin family of proteins [67]. hGMAP-210, which plays a role in maintaining the structural integrity of the Golgi apparatus, binds to the Golgi apparatus through its NH2 terminus and interacts directly with microtubules through its COOH-terminal domain [68]. hGMAP-210 acts at the crossroads between the anterograde and retrograde transport, connecting the ER to the Golgi apparatus. The overexpression of hGMAP-210 can block both anterograde and retrograde transport between the ER and the Golgi apparatus, and significantly alter the morphology of the Golgi complex, resulting in the accumulation of vesicles to form large clusters [69]. In contrast to previous studies, Sato et al. demonstrated that the knockdown of hGMAP-210 results in the Golgi apparatus breakage and significant densification. This may be due to differences in knockdown efficiency. Additionally, they found that hGMAP-210 acts as a Golgi vesicle tether in vivo [70]. Research has demonstrated that hGMAP-210 plays a crucial role in maintaining the Golgi band around the centrosome through its interactions with the Golgi membrane and γ-tubulin [71]. Additionally, hGMAP-210 is necessary for the efficient glycosylation and cellular
Comments (0)