
Remember me



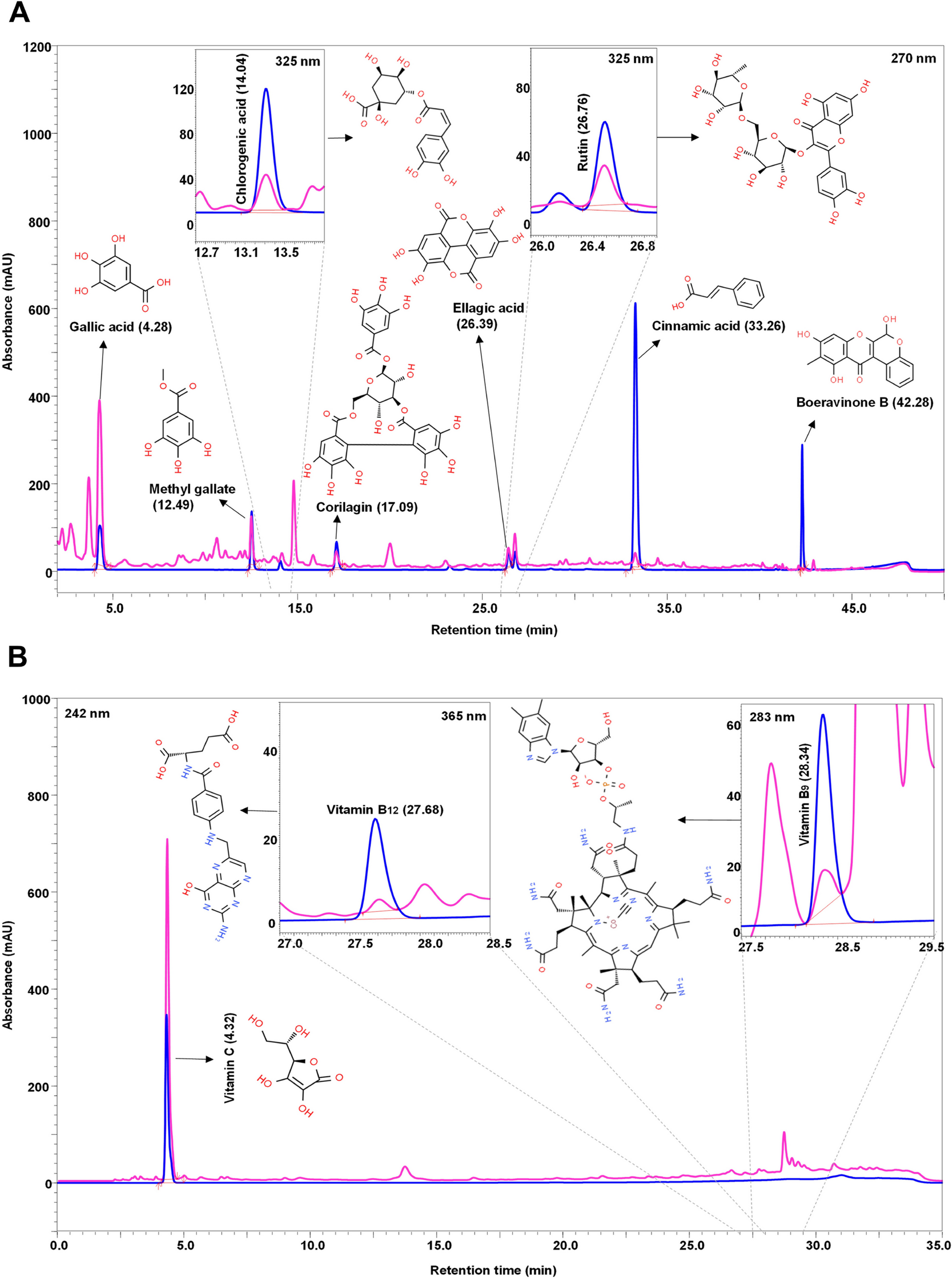
4T1 cells purchased from Procell Life Science&Technology Co.,Ltd.(China) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and incubated at 37 °C in an atmosphere of 5% CO2 air.
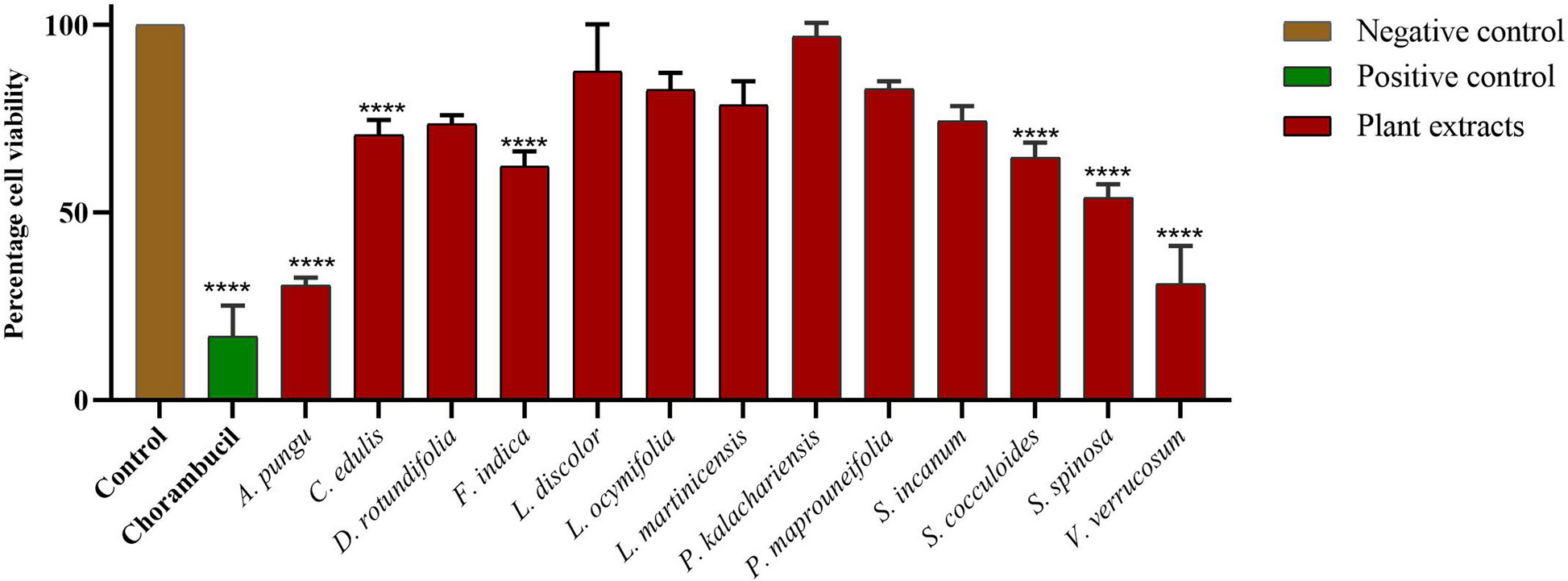
Cell viability assay4T1 cell viability was detected by the 3-(4,5-dimethylthiazol-2-yi(2,5-diphenyltetrazolium bromide (MTT) assay. Each well of 96-well plates was seeded with 5 × 103 cells and incubated overnight. The cells were then exposed to a series of concentrations of shikonin (Desite, China) ranging from 10 to 0.039 µg/mL. After 48 h of treatment, 20 µL of MTT reagent (Solarbio, China) at a concentration of 5 mg/mL was added to each well. After incubation at 37 °C for 4 h, the supernatant was removed. The insoluble formazan that had been formed by viable cells was resuspended in 150 µL dimethylsulfoxide (DMSO; Yuanye, China) and the absorbance was determined using a microplate reader (Thermo Fisher Scientific, USA). The experiment on the effect of shikonin on non-cancerous cells was included in the supporting information.
For in vitro experiments, shikonin was dissolved in DMSO to produce a stock solution of 10 mg/mL, which was diluted as required in cell culture medium supplemented with 10% fetal bovine serum.
Cell apoptosis assayApoptosis was detected by flow cytometry. Each well of six-well plates was seeded with 2 × 105 cells and incubated overnight. The cells were then treated with 200 ng/mL shikonin (low-dose shikonin group, LD-SK group) or 800 ng/mL shikonin (high-dose shikonin group, HD-SK group). Cells in the negative control group were treated with PBS (Procell, China; PBS group). Cells in the positive control group were treated with 400 ng/mL doxorubicin hydrochloride (Meilunbio, China; DOX group). After 48 h of treatment, the cells were collected and stained with Annexin V-Cy5 (BioVison, USA) and 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI; Meilunbio, China) at 4℃ for 30 min, followed by flow cytometry analysis (Beckman Coulter, USA).
ROS assayIntracellular ROS levels were measured by flow cytometry. Cells at logarithmic growth stage were inoculated (3 × 105 cells) in wells of six-well plates and incubated for 24 h. The cells were then treated to yield the aforementioned PBS, DOX, LD-SK, and HD-SK group. After 24 h of treatment, the ROS fluorescent probe dihydrorhodamine 123 (Meilunbio, China) was added and then incubated at 37℃ for a further 4 h in the dark. After incubation, elution with PBS was performed three times, and fluorescence intensity was detected by flow cytometry.
Mitochondrial activity assayThe mitochondrial activity was detected using flow cytometry. Each well of six-well plates was seeded with 2 × 105 cells and incubated for 24 h. The cells were then treated to establish the aforementioned PBS, DOX, LD-SK, and HD-SK groups. After 48 h of treatment, the cells were collected and stained with JC-10 cationic and lipophilic dye (BIOBYING, China) at 37℃ for 2 h. After staining, cells were washed and resuspended in PBS. Mitochondrial activity was detected by flow cytometry.
Calreticulin (CRT) exposure assayCRT exposure was quantified by flow cytometry. Cells at the logarithmic growth stage were inoculated (2 × 105 cells) in wells of six-well plates and incubated for 24 h. The cells were then treated to establish the aforementioned PBS, DOX, LD-SK, and HD-SK groups. After 48 h of treatment, the cells were collected and incubated with the CALR Polyclonal Antibody (Elabscience, China) at 37℃ for 1 h. The cells were washed with PBS and incubated with goat Anti-Rabbit IgG H&L/Cy5 (Bioss, China) at 37 °C in the dark for 1 h, washed, and resuspended in PBS. Flow cytometry was used for the quantitative assay of CRT exposure of 4T1 cells.
In vivo antitumor activityTwenty-four specific pathogen-free healthy female BALB/c mice (6–8 weeks old, 16–20 g in weight) were obtained from the Laboratory Animal Center, Guangzhou University of Chinese Medicine (License Number: SYXK (Yue) 2018-0034). The mice were raised in a temperature-and humidity-controlled pathogen-free room under a 12 h:12 h light/dark cycle with ad libitum access to food and water. All animal experiments were approved by the Ethics Committee of Sci-tech Industrial Park, Guangzhou University of Chinese Medicine (serial number: PZ22069).
After being anesthetized by isoflurane (Ruiwode, China) inhalation (1.5% in air), the mammary fat pads of 18 mice were injected with 4T1 cells (5 × 105 cells in 50 µL PBS per mouse) using sterile insulin syringe (Kindly, China). Animals were randomized (n = 6) and then received intravenous injection of shikonin (10 mg/kg, dissolved in PBS containing 1% DMSO and 3% Tween 80), DOX (2.5 mg/kg, dissolved in PBS), or an equal volume of PBS every other day for 14 days (Fig. 2). The tumor volume of mice were measured every other day (tumor volume = length×1/2 width2). Animals were euthanized by isoflurane inhalation (3% in air) and cervical dislocation after treatment. The tumor tissues and spleens were collected.
Fig. 2
Schematic diagram of in vivo experimental procedure
Spleen immune cells detectionSpleen immune cells were analyzed by flow cytometry. Spleens were washed with pre-cooled PBS, ground, and passed through a 200 sieve to purify the spleen grinding liquid. The collected cell suspension was centrifuged at 3000 rpm for 5 min. After removing the supernatant, 1 mL of diluted modified red blood cell lysis buffer (Meilunbio, China) was added to the cell precipitate and the cells were lysed at room temperature for 10 min. When a white flocculent precipitate was visible to the naked eye, 1 mL of PBS was added to terminate lysis, and each sample was centrifuged after filtering. The precipitate was resuspended with PBS to form a single cell suspension of spleen cells. The spleen single cell suspension was divided into two equal portions. One portion was incubated with Elab Fluor® Violet 450 Anti-Mouse CD45 Antibody[30-F11] (Elabscience, China), Elab Fluor® Red 780 Anti-Mouse CD3 Antibody[17A2] (Elabscience), APC Anti-Mouse CD4 Antibody[GK1.5] (Elabscience, China) and fluorescein isothiocyanate (FITC) Anti-Mouse CD8a Antibody[53 − 6.7] (Elabscience) diluted in cell staining buffer (Elabscience, China) at 4 °C in the dark for 30 min. The other portion was incubated with Elab Fluor® Violet 450 Anti-Mouse CD45 Antibody[30-F11], Elab Fluor® Red 780 Anti-Mouse CD3 Antibody[17A2], APC Anti-Mouse CD4 Antibody[GK1.5], and PerCP/Cyanine5.5 Anti-Mouse CD25 Antibody[PC-61.5.3] (Elabscience, China) diluted in cell staining buffer at 4 °C in the dark for 30 min. Fixed membrane breaking and nucleation breaking solutions (Biolegend, USA) were used to disrupt cell membranes and nuclei, respectively, according to the specifications to facilitate staining with phycoerythrin (PE) Anti-Mouse Foxp3 Antibody [3G3] (Elabscience, China). Finally, flow cytometry was used to examine the samples, and FlowJo _V10 software was used for data analysis.
Statistical analysisSPSS Statistics version 22.0 (IBM, USA) was used for the statistical analysis of data. Data are presented as mean ± standard error. One-way analysis of variance (ANOVA) was used for the measurement of multiple groups conforming to a normal distribution and homogeneity of variance. Welch’s ANOVA was used for the measurement data of multiple groups that conformed to a normal distribution but did not conform to homogeneity of variance. Kruskal-Wallis test was used for the measurement data of multiple groups that did not conform to a normal distribution. The test level of the above statistical results used P < 0.05 as the standard of significant difference and P < 0.01 as the standard of very significant difference.
Comments (0)