
Remember me




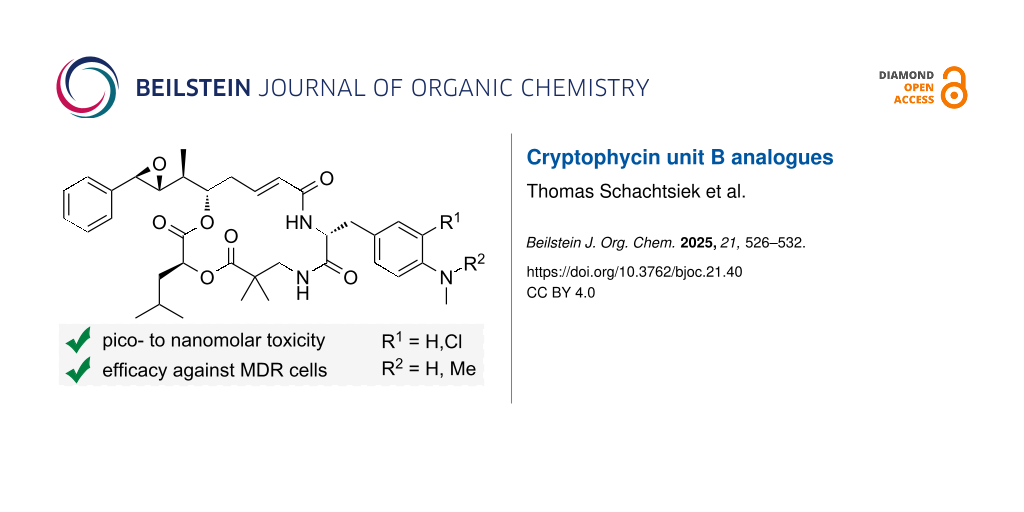
Cryptophycins emerged as highly potent cytotoxins for the use in targeted cancer therapy . Originally discovered over three decades ago by isolation from cyanobacteria , their extraordinarily high cytotoxicity was apparent and attracted attention, not least because of their still high efficacy against multidrug-resistant (MDR) cells . Cryptophycin-52, a synthetic development candidate by Eli Lilly based on the initially discovered cryptophycin-1, failed in clinical studies as a cytotoxic drug on its own due to severe side effects . However, the embedment of cryptophycins as payloads in drug conjugates, increases specificity and minimises adverse effects . Drug conjugates usually consist of three units, where the payload is connected to the homing device by a linker. For covalent attachment of the drug to the linker a suitable functional group is needed such as an amino, hydroxy, carboxy or sulfhydryl group . Since the cryptophycins’ discovery, considerable efforts were made for the establishment of structure–activity relationship (SAR) studies between derivatisations of all four units (A to D) (Figure 1A) of cryptophycin-52 analogues with functional groups . While many modifications of cryptophycin decrease the cytotoxicity drastically, some viable attachment points for conjugation were found, mainly including the para-phenyl position of unit A or derivatisation of unit A’s epoxide moiety into a halohydrin (-glycinate) . Cryptophycins modified at the α-position of unit C and, most recently, various derivatives with modifications in unit D were established as potent and conjugable payloads by our group. Modifications of unit B with conjugation handles are scarcely explored and mainly include the exchange of the para-methoxy group (Figure 1B). The sole exchange of the para-methoxy group of cryptophycin-52 (IC50 = 22 pM) by a hydroxy group reduces the cytotoxicity by approximately only one order of magnitude (IC50 = 0.52 nM) when tested on CCRF-CEM T lymphoblasts . Loss of the meta-chloro substituent shows a similar trend. Functionalisation of the hydroxy group with ethylene glycol residues further decreases cytotoxicity, whereby this effect increases with increasing chain length . The exchange by an amino group showed a similar trend, however, N,N-dimethylation of the amino group again increased cytotoxicity significantly . Nevertheless, this cryptophycin might only be conjugated via an ammonium-based self-immolative linker . We envisioned the synthesis of, firstly, m-chloro-p-(methylamino) derivative 1 to dissect the structure–activity relationship between the primary (IC50(CCRF-CEM) = 0.58 nM), secondary, and tertiary amine (IC50(CCRF-CEM) = 54 pM) derivatives . Secondly, p-(dimethylamino) derivative 2 was synthesised to investigate the effect of N-alkylation on the non-chlorinated unit B derivatives.
![[1860-5397-21-40-1]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-21-40-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: A: Structure of cryptophycin-52. B: Cryptophycin-52 derivatives modified with conjugation handles in unit B. IC50 values given against (a) CCRF-CEM and (b) KB-3-1 cell lines. C: Cryptophycin-52 derivatives synthesised in this work.
Results and DiscussionFor the synthesis of unit B derivatives with amino groups instead of the naturally occurring methoxy group ᴅ-phenylalanine served as the fundamental substrate (Scheme 1). Nitration followed by methyl ester formation and N-Boc-protection provided nitroarene 5 in 40% yield over three steps. Reduction of the nitro group was performed with Pd/C and hydrogen to obtain aniline 6 in 98% yield, which served as a precursor for mono- and dimethylated unit B derivatives 7 and 8, respectively. While dimethylaniline 8 was obtained in good yield of 61% through reductive amination with excess formaldehyde and NaBH3CN as reductant, the selective installation of only one methyl group, providing monomethyl aniline 7, proved to be more troublesome. Either reductive amination using the same protocol, but under strict control of equivalents and pH, or Leuckart–Wallach-like reaction with ammonium formate and Pd/C provided monomethylaniline 7 in 37% and 35% yield, respectively. The absolute structure of monomethylaniline 7 was confirmed by single-crystal X-ray diffraction measurements (Figure 2). Compound 7 crystallised in the monoclinic space group P21 with R1 = 0.0285 clearly showing the expected (R)-configuration with a Flack parameter of −0.07(6).
![[1860-5397-21-40-i1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-40-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of modified unit B derivatives. a) HNO3, H2SO4, 0 °C, 5 h, 48% (isolated as monohydrate); b) SOCl2, MeOH, 0 °C, 90 min, then reflux, 17 h, 95%; c) Boc2O, NEt3, MeCN, H2O, rt, 22 h, 88%; d) H2, Pd/C, MeOH, rt, 25 h, 98%; e) either: formalin, NaBH3CN, HOAc, MeOH, rt, 20 min, 37%, or: formalin, (NH4)HCO2, Pd/C, MeOH, rt, 20 min, 35%; f) N-chlorosuccinimide, MeCN, reflux, 16 h, 77%; g) AllocCl, NaHCO3, CHCl3, H2O, rt, 5 h, 89%; h) LiOH·H2O, MeOH, tetrahydrofuran, H2O, 0 °C to rt, 3 h, 99%; i) formalin, NaBH3CN, MeCN, rt, 55 min, 61%; j) 1. TFA, CH2Cl2, 0 °C, 1.5 h; 2. acryloyl chloride, NEt3, CH2Cl2, 0 °C to rt, 19 h, 63% over two steps; k) LiOH·H2O, MeOH, tetrahydrofuran, H2O, 0 °C, 3.5 h, 82% with unconverted 12 (18 mol %).
![[1860-5397-21-40-2]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-21-40-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of Boc-ᴅ-Phe(4-NHMe)-OMe 7 as determined by single-crystal X-ray diffraction measurements. Thermal ellipsoids depicted at 50% probability.
Starting from monomethylaniline derivative 7, the synthesis of the final building block was finalised by chlorination , Alloc protection and saponification to obtain free acid 11. Exchange of the Boc protecting group of dimethylaniline 8 to an acryloyl substituent and subsequent saponification furnished dimethylamine building block 13.
For the macrocycle assembly, especially the ring closure, we decided on two different routes. While the cryptophycin containing a dimethylamino motif did not require an additional protecting group, ring closure was performed through alkene cross metathesis, which has been accomplished reliably and with good yields for other cryptophycins . However, for the synthesis of a cryptophycin with a monomethylated amino group in unit B a suitable protecting group, i.e., allyloxycarbonyl (Alloc), must be used. Since the presence of this allylic double bond would most likely interfere with a clean reaction outcome after alkene cross metathesis, we decided for a more classical ring-closure strategy through macrolactamisation .
The syntheses of required unit A , C , and D building blocks were accomplished as described previously. tert-Butyl-protected leucic acid 14 and Fmoc-β-aminopivalic acid (15) were connected by Steglich esterification (Scheme 2) and after cleavage of the tert-butyl ester group coupled to either one of the two unit A precursors 18 and 19 by Yamaguchi esterification. CDA fragments 20 and 21 were connected to unit B derivatives 11 and 13 through amidation which provided seco-cryptophycins 22 and 23 in yields of 49% and 38%, respectively. Starting from seco-cryptophycin 22 acidolysis was followed by macrolactamisation to obtain diol 24, albeit in poor yield of 11%. LC–MS analyses revealed that the low yield cannot be attributed to either incomplete acidolysis or incomplete conversion of the deprotected seco-cryptophycin during macrolactamisation. Rather, LC–MS analyses indicate the major presence of a fully deprotected and trifluoroacetylated seco-cryptophycin, most likely a TFA ester of one of the free hydroxy groups, a finding which might reason the comparably low yields (21–25%) of structurally similar unit B analogues reported earlier . Contrary, diol 25 was obtained through Grubbs metathesis and subsequent acetonide cleavage in a superior yield of 76%.
![[1860-5397-21-40-i2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-40-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of cryptophycin diols 24 and 25. a) EDC·HCl, DMAP, NEt3, CH2Cl2, 0 °C to rt, 22 h, 60%; b) TFA, CH2Cl2, 0 °C, 4 h, 100%; c) 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, THF, 0 °C or 0 °C to rt, 3h, tetrahydrofuran, 77% (for 20)/88% (for 21); d) 1. piperidine, N,N-dimethylformamide, rt, 1.5 h; 2. unit B (11 for 20 and 13 for 21), HOAt, NEt3, EDC·HCl, CH2Cl2, 0 °C to rt, 18 h, 49% (for 22)/38% (for 23); e) 1. TFA, CH2Cl2, 0 °C to rt, 2.5 h; 2. HATU, HOAt, N,N-dimethylformamide 0 °C for 2 h and at rt for 20 min, 11%; f) Grubbs II catalyst, CH2Cl2, reflux, 3 h, 84%; g) TFA, H2O, CH2Cl2, 0 °C to rt, 5 h, 91%. EDC·HCl = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride; DMAP = 4-(dimethylamino)pyridine; TFA = trifluoroacetic acid; HOAt = 1-hydroxy-7-azabenzotriazole; HATU = 1-[bis(dimethylamino)methyliumyl]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate.
The finalising steps to obtain epoxides 26 and 2 (Scheme 3) were a diol–epoxide transformation , including firstly the formation of a cyclic orthoester, secondly the formation of a bromohydrin formate, and thirdly ring closure to obtain the epoxides 26 and 2 in 89% and 14% yield over three steps each, respectively.
![[1860-5397-21-40-i3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-40-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Three-step diol–epoxide transformation starting from diols 24 and 25. a) (MeO)3CH, pyridinium p-toluenesulfonate, CH2Cl2, rt, 3 h; b) AcBr, CH2Cl2, rt, 3–4 h; c) K2CO3, MeOCH2CH2OMe, HOCH2CH2OH, rt, 5 min, 89% (for 26)/14% (for 2), over three steps each; d) Pd(PPh3)4, morpholine, CH2Cl2, rt, 90 min, 74%.
While the synthesis of tertiary amine 2 was finished, final Alloc cleavage from 26 under Tsuji–Trost-like conditions provided secondary amine 1 in 74% yield.
The cytotoxicity of Alloc-protected cryptophycin 26 and both free amines 1 and 2 were tested against the human cervix carcinoma cell line KB-3-1 and its MDR subclone KB-V1 (Table 1) .
Table 1: IC50 values of synthesised cryptophycins against KB-3-1 and KB-V1 cell lines.
IC50 [nM] KB-3-1 KB-V1 26 374 >1000 1 0.313 7.76 2 6.36 218The m-chloro-p-(methylamino) derivative 1 showed high cytotoxicity with IC50 (KB-3-1) = 313 pM, which is perfectly between the values of the primary (IC50 (CCRF-CEM) = 580 pM) and tertiary (IC50 (CCRF-CEM) = 54 pM) amine derivatives (Figure 1B) , showing a strict increase in cytotoxicity with increasing degree of aniline methylation. The same trend was also observed for unit D derivatives modified with amino groups and likely hints to favoured hydrophobic interactions around the binding pocket at unit B and D. In comparison, the cytotoxicity of Alloc-protected amine 26 decreased significantly (IC50 (KB-3-1) = 374 nM), which could be due to higher steric bulk as this is known to be less well tolerated; cf. unit B p-alkyloxy derivatives (Figure 1B) . Tertiary amine 2 showed IC50 (KB-3-1) = 6.36 nM which is similar to its non-alkylated congener (IC50 (CCRF-CEM) = 10.11 nM) but about two orders of magnitude higher than its chlorinated derivative (IC50 (CCRF-CEM) = 54 pM) (Figure 1B) . Notably, both free amines still show nanomolar activity against the MDR cell line KB-V1 (Table 1) with resistance factors FR (being the ratio between IC50 (KB-V1) and IC50 (KB-3-1)) of 25 (1) and 34 (2), thus being comparably much more potent against KB-V1 cells than for example the unit D analogues (FR ≈ 103) .
ConclusionIn summary, we synthesised two new conjugable cryptophycin analogues 1 and 2 with amino groups in unit B, by utilising either macrolactamisation or Grubbs metathesis for ring closure. These synthetic routes should generally allow access to diverse other unit B cryptophycin analogues. Both cryptophycins 1 and 2 showed high cytotoxicity with 313 pM (1) and 6.36 nM (2) and outstandingly low resistance factors. Furthermore, the new cryptophycin 1 confirms the correlation between degree of alkylation and cytotoxicity of m-chloro-p-amino unit B derivatives. Since MDR is responsible for over 90% of deaths in cancer patients and MDR-selective therapeutics are lacking , we expect that cryptophycins 1 and 2 will be considered as viable cytotoxins for the use in targeted tumour therapy, especially against MDR cancers.
© 2025 Schachtsiek et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.
Comments (0)