
Remember me



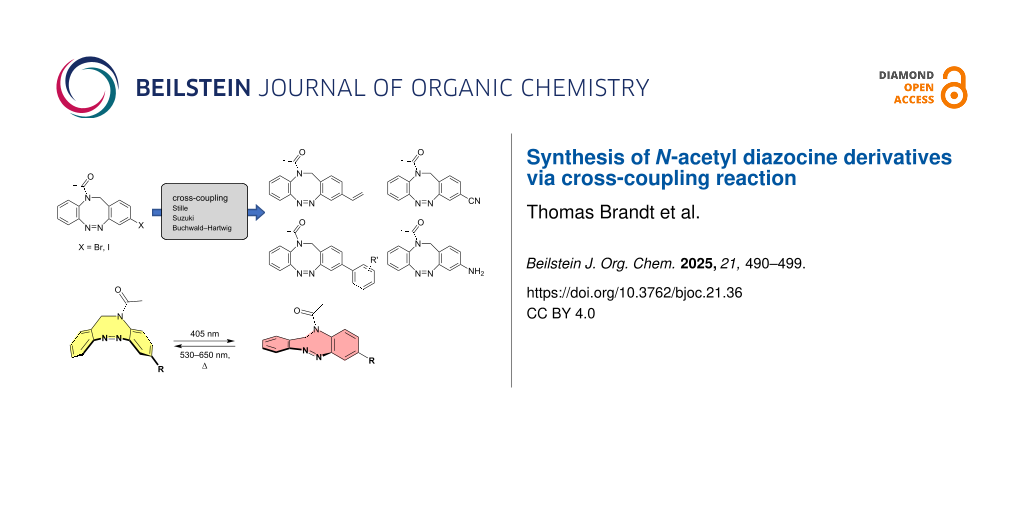

Diazocines are frequently used photoswitches with superior photophysical properties. The parent ethylene-bridged diazocine shows excellent switching photoconversion between the Z and the E configurations (Γ(Z → E)385nm = 92% and Γ(E → Z)520nm > 99% in n-hexane) due to well-separated n–π*-transitions in the visible part of the electromagnetic spectra . Moreover, the ethylene bridge creates a cyclic 8-membered core, inverting the thermodynamically stability in favor of the Z boat conformation compared to parent azobenzene, which has a stable E configuration . Preceding studies including azobenzene-based photopharmacophores showed that, in most cases, the sterically demanding Z configuration is biologically inactive, while the stretched E configuration is biologically active . Because of the inverted thermodynamic stability compared to azobenzene, the stable Z configuration of the diazocine can be administered and subsequently activated with light at the site of illness with high spatiotemporal resolution. Thus, collateral damage in the surrounding healthy tissue can be avoided. In addition, the quantitative thermal back-isomerization from the active E to the inactive Z configuration prevents contamination and accumulation in the environment after excretion . These superior properties of diazocines have been exploited in several applications such as the control of protein folding by implementation as cross-linkers between protein side chains or in peptide backbones , as photoswitchable neurotransmitters or as switching units for potential dependent potassium channels . Compared to the Z → E conversion rate of 92% (in n-hexane) of the parent diazocine the conversion in water/DMSO mixtures is decreasing with increasing water concentration (73% in water/DMSO 9:1) . Moreover, the parent diazocine is insoluble in water (precipitation in water/DMSO > 9:1). Substitution with polar substituents such as CH2NH2 provides water solubility, however, it does not restore the high Z → E conversion rates of the parent system in organic solvents, which is a disadvantage, since biochemical reactions usually take place in aqueous environments . The substitution of one CH2 group in the CH2CH2 bridge by N–C(=O)–CH3 leads to an intrinsic water solubility of the N-acetyl diazocine 1 (Figure 1) . Furthermore the photoconversion of 1 shows no significant drop in pure water in contrast to the solubilized parent diazocine. These superior properties make N-acetyl diazocine 1 an ideal candidate for application in the field of photopharmacology especially in aqueous environments . The same applies for the quantum yields. While the quantum yields ΦZ→E and ΦE→Z of the parent diazocine drop significantly in aqueous media, the corresponding quantum yields of the N-acetyl diazocine stay the same or even slightly improve (Table 1). In general, the quantum yields of parent diazocine and its’ nitrogen bridged derivative exceed the quantum yields of other molecular photoswitches like azobenzenes, spiropyranes and diarylethenes in organic solvents .
Table 1: Quantum yields of N-acetyl diazocine 1 in organic and aqueous media compared to parent diazocine .
parent diazocine N-acetyl diazocine solvent ΦZ→E (385 nm) ΦE→Z (520 nm) ΦZ→E (400 nm) ΦE→Z (520 nm) acetone 0.72 0.90 0.48 0.85 acetonitrile 0.43 0.56 0.48 0.79 MeCN/H2O 9:1 0.37 0.56 – – H2O – – 0.51 0.85There are two strategies of applying diazocines in photopharmacology. The first one exploits the structural similarity of the tricyclic diazocine framework to the tricyclic structure of, e.g., tetrahydrodibenzazocines and tetracyclic steroid scaffolds such as 17β-estradiol , where the diazocine core mimics the framework of the bioactive compound (Figure 1a). The other option is to attach the diazocine photoswitch as a substituent (appendix) to the biologically active molecule (Figure 1b) . The art of designing a photoswitchable drug is to place the switch at a position in the pharmacophore that allows switching of the biological effect by irradiation with light without greatly reducing the overall activity by unselective interference with the inhibitor–receptor interaction. This is a difficult task because the design of a photoswitchable agent usually starts with a known, non-switchable drug or a known biological molecule, which is already carefully "optimized" either by pharmaceutical industry or by nature. Hence, there is a high risk that any change in structure will also lead to a reduction in efficiency. In any case the light-induced geometry change via isomerization should selectively control the interaction between the inhibitor and the receptor .
![[1860-5397-21-36-1]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-21-36-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Structural similarity of N-acetyl diazocine 1 with known 17βHSD3-inhibitor tetrahydrodibenzazocine (THB) and parent diazocine with steroid scaffolds . b) Parent diazocine attached to glutamate to form a photoswitchable neurotransmitter .
Currently there is only one example reported in the literature for the incorporation of N-acetyl diazocines into biologically active molecules . As a starting point for further derivatization, the synthesis and characterization of monohalogenated N-acetyl diazocines 2 and 3 (Figure 1) have been performed . Unfortunately, diazocines in general, and N-acetyl diazocines in particular cannot be derivatized by electrophilic aromatic substitution. Substituents such as halogen atoms must be introduced into the N-acetyl diazocine structure during the synthesis of the building blocks. In the present work we start from mono- and dihalogenated N-acetyl diazocine 2–4 (Figure 2) and focus on the further derivatization via cross-coupling reactions and the synthesis of a new dihalogenated N-acetyl diazocine 4 (Figure 2).
![[1860-5397-21-36-2]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-21-36-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The halogen-substituted N-acetyl diazocines 2–4 were used as the starting compounds for further derivatization via Pd-catalyzed cross-coupling reactions. Solutions of the Z isomers are yellow. The E isomers are red.
Results and DiscussionThe monosubstituted N-acetyl diazocines 2 and 3 were synthesized according to the procedure published by our group recently . The synthesis of disubstituted compound 4 followed the same procedure except the preparation of the aminoaniline building block tert-butyl (2-amino-5-bromophenyl)carbamate (5), which was prepared by Boc-protection of the 5-bromo-2-nitroaniline (6) and subsequent reduction of the nitro group (see Supporting Information File 1, section II.1).
Cross-coupling reactionsStille cross-coupling reactions were performed by an organic halide reacting with an organotin compound. A great advantage of the used organostannanes is the easy accessibility, and their high air and moisture stability, so that usually a wide range of functional groups can be introduced under mild conditions . Nevertheless, the arylation of monohalogenated N-acetyl diazocines via Stille coupling in our case gave unsatisfying results (Table 2). Reactions with tetrakis(triphenylphosphine)palladium(0) as catalyst resulted in no product 7 formation. Bis(tri-tert-butylphosphine)palladium(0) as catalyst gave rise to the product in very low yields independent from the used halogenated diazocine. In contrast to other cross coupling reactions described in this work, most of the starting material decomposed during the reaction and could not be re-isolated.
Table 2: Reaction conditions of the arylation of halogenated N-acetyl diazocines via Stille coupling reaction. Equivalents are normalized to the used amount of N-acetyl diazocine starting material.
![[Graphic 1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i4.svg?max-width=637&scale=1.0) cat. system
organotin compound
conditions
yield
Pd(OAc)2 (0.1 equiv) + PPh3 (0.8 equiv)
Ph3SnCl (1 equiv)
dry DMF, N2, 100 °C, 16 h
–
Pd(t-Bu3P)2 (0.1 equiv)
Ph3SnCl (1 equiv)
dry THF, N2, 65 °C, 16 h
X = Br, 10%
cat. system
organotin compound
conditions
yield
Pd(OAc)2 (0.1 equiv) + PPh3 (0.8 equiv)
Ph3SnCl (1 equiv)
dry DMF, N2, 100 °C, 16 h
–
Pd(t-Bu3P)2 (0.1 equiv)
Ph3SnCl (1 equiv)
dry THF, N2, 65 °C, 16 h
X = Br, 10%
In contrast, the vinylation of diazocines 2 and 3 provides good yields of 65% and 71%, respectively, for the vinyl N-acetyl diazocine 8 (Table 3). An alternative way of vinylating N-acetyl diazocines is the Pd-catalyzed vinylation with polyvinylsiloxanes and TBAF as activating agent following the method by Denmark et al. giving rise to even higher yields of 74% for bromine 2 and 78% for iodo starting material 3 (Table 3) .
Table 3: Vinylation of halogenated N-acetyl diazocines via Pd-catalyzed coupling reactions. Equivalents are normalized to the used amount of N-acetyl diazocine starting material.
![[Graphic 2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i5.svg?max-width=637&scale=1.0) cat. system
reaction partner or additive
conditions
yield
Pd(OAc)2 (0.1 equiv) + PPh3 (0.8 equiv)
tributylvinyltin (1 equiv)
dry DMF, N2, 100 °C, 16 h
X = Br, 65%
cat. system
reaction partner or additive
conditions
yield
Pd(OAc)2 (0.1 equiv) + PPh3 (0.8 equiv)
tributylvinyltin (1 equiv)
dry DMF, N2, 100 °C, 16 h
X = Br, 65%
aD4V: 1,3,5,7-tetramethyl-1,3,5,7-tetravinylcyclotetrasiloxane.
To overcome the problems of poor yields in the arylation of N-acetyl diazocines via Stille coupling we used Suzuki–Miyaura reactions of the diazocines 2 and 3 with different arylboronic acids . There are several examples of last-step modifications of azobenzenes via Suzuki–Miyaura reactions in the current literature, which indicate that the reaction conditions are compatible with azo groups . Suzuki–Miyaura reaction of 2 and 3 with different phenylboronic acids resulted in the formation of the corresponding arylated N-acetyl diazocines 7, and 9–13 in yields from 68 to 88% (Table 4). The yields increased slightly if boronic acids with electron-withdrawing groups were used. An influence of bulky substituents like carboxyl groups in ortho-position of the phenylboronic acids on the reaction was not observed. The synthesis of N-acetyl diazocines connected to heteroaromatic aromatic systems 14–16 was less successful. The pyridine-substituted N-acetyl diazocine 14 was formed in yields of 7% or 19% while furan- 15 and thiophene-substituted N-acetyl diazocine 16 could not be obtained. The reaction with benzylboronic acid gave the corresponding N-acetyl diazocine 17 (45%, Table 4). Interestingly, this reaction only took place if brominated N-acetyl diazocine 2 was used as starting material although iodoaryl compounds are in general more reactive . The reaction of halogenated N-acetyl diazocines 2 and 3 with bis(pinacolato)diboron did not lead to the formation of the pinacolborane-substituted N-acetyl diazocine 18. Accordingly, the Suzuki–Miyaura reaction with inversed roles between N-acetyl diazocine boronic acid pinacol ester and aryl or alkyl halides could not be investigated.
Table 4: Derivatization of halogen-substituted N-acetyl diazocines via Suzuki–Miyaura reaction. Equivalents are normalized to the used amount of N-acetyl diazocine starting material.
![[Graphic 3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i6.svg?max-width=637&scale=1.0) boronic acid
product
yield
boronic acid
product
yield
![[Graphic 4]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i7.svg?max-width=637&scale=1.0)
![[Graphic 5]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i8.svg?max-width=637&scale=1.0)
![[Graphic 6]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i9.svg?max-width=637&scale=1.0)
![[Graphic 7]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i10.svg?max-width=637&scale=1.0)
![[Graphic 8]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i11.svg?max-width=637&scale=1.0)
![[Graphic 9]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i12.svg?max-width=637&scale=1.0)
![[Graphic 10]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i13.svg?max-width=637&scale=1.0)
![[Graphic 11]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i14.svg?max-width=637&scale=1.0)
![[Graphic 12]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i15.svg?max-width=637&scale=1.0)
![[Graphic 13]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i16.svg?max-width=637&scale=1.0)
![[Graphic 14]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i17.svg?max-width=637&scale=1.0)
![[Graphic 15]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i18.svg?max-width=637&scale=1.0)
![[Graphic 16]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i19.svg?max-width=637&scale=1.0)
![[Graphic 17]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i20.svg?max-width=637&scale=1.0)
![[Graphic 18]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i21.svg?max-width=637&scale=1.0)
![[Graphic 19]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i22.svg?max-width=637&scale=1.0)
![[Graphic 20]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i23.svg?max-width=637&scale=1.0)
![[Graphic 21]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i24.svg?max-width=637&scale=1.0)
![[Graphic 22]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i25.svg?max-width=637&scale=1.0)
![[Graphic 23]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i26.svg?max-width=637&scale=1.0)
![[Graphic 24]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i27.svg?max-width=637&scale=1.0)
![[Graphic 25]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i28.svg?max-width=637&scale=1.0)
aThe reaction was carried out in dry DMF at 100 °C because no reaction took place if the Suzuki–Miyaura standard procedure was applied.
The Buchwald–Hartwig amination is a versatile and powerful tool for C–N bond formation and widely applied in the synthesis of new pharmaceutical substances . Furthermore, azobenzenes , as well as diazocines , have been derivatized via Buchwald–Hartwig amination. The Buchwald–Hartwig amination of halogenated N-acetyl diazocines according to the procedure of Maier et al. with tert-butyl carbamate resulted in the formation of Boc-protected amino-substituted N-acetyl diazocine 19 in a yield of 72%. However, the reaction only took place if iodo N-acetyl diazocine 3 was used as starting material. Using diphenylamine as a more electron-rich amine resulted in the formation of diphenylamino-substituted N-acetyl diazocine 20 in a significantly lower yield of 25% starting from the bromide 2 and 47% starting from the iodo precursor 3 (Table 5).
Table 5: Derivatization of halogenated N-acetyl diazocines 2 and 3 via Buchwald–Hartwig amination. Equivalents are normalized to the used amount of N-acetyl diazocine starting material.
![[Graphic 26]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i29.svg?max-width=637&scale=1.0) nitrogen compound
product
yield
nitrogen compound
product
yield
![[Graphic 27]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i30.svg?max-width=637&scale=1.0)
![[Graphic 28]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i31.svg?max-width=637&scale=1.0)
![[Graphic 29]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i32.svg?max-width=637&scale=1.0)
![[Graphic 30]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i33.svg?max-width=637&scale=1.0)
Deprotection of carbamate 19 with trifluoroacetic acid provided the corresponding amino-substituted N-acetyl diazocine 21 (Scheme 1).
![[1860-5397-21-36-i1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of amino-N-acetyl diazocine by deprotection of the carbamate.
Another option for carbon–heteroatom bond formation reactions are copper-catalyzed Ullmann-type reactions, which have already been applied to the parent diazocine . The attempted synthesis of azide-functionalized N-acetyl diazocine 22 under the conditions described by Hugenbusch et al. showed no product formation and only starting material was isolated (Scheme 2).
![[1860-5397-21-36-i2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction conditions for the attempted Ullmann-type reaction with sodium azide.
The palladium-catalyzed introduction of cyano groups under mild conditions in analogy to Iqbal et al. gave the cyano-substituted N-acetyl diazocine 23 in yields of 61% from bromide 2 and 81% from iodide 3 (Scheme 3). Nitriles are a good starting point for further functional group interconversions .
![[1860-5397-21-36-i3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-21-36-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction conditions for the palladium-catalyzed introduction of a nitrile functionality.
Photochemical characterizationWith these new N-acetyl diazocine derivatives at hand we turned towards the photochemical characterization, in particular to gain insight into the effects of different substituents on UV spectra and switching behavior. For the determination of the n–π*-absorption maxima of the E and Z isomers 250 µM solutions of each compound in acetonitrile were prepared and measured at 25 °C. All compounds (4, 7–14, 17, 19–21, 23) exhibit an n–π*-transition at approximately 400 nm, matching the n–π*-transition of unsubstituted N-acetyl diazocine 1 (Table 6). Irradiation with light of 405 nm gives the metastable E isomers with photoconversion yields of 76–85% due to a very good separation of the n–π*-transitions. N-Acetyl diazocine derivatives containing electron-deficient groups (like 4, 10, 12, 14 and 23) show slightly but not significantly higher Z→E conversion rates than the other coupling products presented in this work. The nitrogen-substituted derivatives 19–21 show significantly lower conversion rates of 41–61%. This behavior has already been observed in other amino-substituted diazocines as well and is probably caused by the overlap of n–π*-transitions of the E and Z isomers and the electron-rich azo group . An almost complete E→Z conversion (>99%) can be achieved by irradiation with light between 520 and 600 nm for all synthesized compounds.
Table 6: Photophysical properties of N-acetyl diazocines 1-4, 7–14, 17, 19–21, and 23 in acetonitrile.
λmax (Z) λmax (E) t1/2 (25 °C) k (25 °C) ΓZ→E (405 nm)a ΓE→Z (530 nm) nm nm min 10−4 s−1 1 397 513 29.5 3.916 88% >99% 2 397 515 30.9 3.744 81% >99% 3 397 517 28.6 4.036 82% >99% 4 396 519 16.9 ± 0.20 6.820 ± 0.096 85% >99% 7 399 516 29.3 ± 0.11 3.944 ± 0.017 79% >99% 8 398 515 29.6 ± 0.16 3.897 ± 0.025 76% >99% 9 394 515 34.2 ± 0.28 3.374 ± 0.032 77% >99% 10 396 516 34.2 ± 0.28 3.382 ± 0.033 82% >99% 11 397 515 33.8 ± 0.24 3.418 ± 0.029 78% >99% 12 396 517 29.8 ± 0.09 3.877 ± 0.014 80% >99% 13 399 517 31.2 ± 0.18 3.688 ± 0.024 77% >99% 14b 396 515 39.9 ± 0.44 2.897 ± 0.038 79% >99% 17 398 515 29.3 ± 0.05 3.944 ± 0.008 82% >99% 19 397 514 30.6 ± 0.05 3.779 ± 0.008 61% >99% 20 389 510 31.5 ± 0.05 3.670 ± 0.008 49% >99% 21 396 511 38.9 ± 0.49
Comments (0)