Study design and participants
This was a prospective, multicenter, randomized, double-blind, parallel-group, placebo-controlled, phase III study to evaluate the efficacy and safety of elobixibat tablet in patients with chronic constipation.
The study planned to enroll eligible male and female patients aged 18 to 65 years (both inclusive), with chronic constipation of at least six months' duration. Patients were diagnosed on the basis of standard symptom-based criteria of fewer than three SBMs per week (defined as bowel movements occurring spontaneously and independently of administration of rescue medication for at least 24 hours), with at least one of the following symptoms during 25% or more of bowel movements: straining at defecation, lumpy or hard stools and sensation of incomplete evacuation. These are included in Rome IV criteria for functional constipation [19].
Patients experiencing constipation needed to have documented results from a colonoscopy or sigmoidoscopy conducted within two years leading up to their screening visit. This requirement was put in place to rule out any organic causes for constipation.
Patients having any cardiovascular, liver, renal or psychiatric disorder or any known adverse drug reaction (ADR) to study drug and excipients were excluded. Pregnant and breast-feeding women were also excluded from our study. The following conditions disqualified patients from participation: prior intestine or rectum surgery (except for a simple appendectomy); prohibited medications, organic disorders of the intestine such as mechanical obstruction; ischemic colitis, inflammatory bowel disease, colorectal cancers and pre-malignant colonic disease (e.g. familial adenomatous polyposis or hereditary non-polyposis colorectal cancer) or other forms of familial colorectal cancer. The requirement for screening colonoscopy was waived for individual patients under 45 years of age, provided they had not experienced alarm symptoms such as weight loss, rectal bleeding or anemia in the past six months.
The study consisted sequentially of a screening period of 14 days, a “pre-treatment” (run-in) period of 14 days followed by the randomized treatment period. The treatment period started with randomization visit (Day 0) and continued for two weeks. Primary efficacy and end of study assessments were performed on Day 14. Patients reporting more than 3 SBM per week on average during pre-treatment period were excluded from the study.
Intervention and randomization
Eligible patients, recruited from 16 clinical sites in India, were randomized using interactive web response systems (IWRS) in 1:1 ratio (either to elobixibat or to placebo treatment groups). Randomized patients took the assigned study drug once per day before breakfast and were monitored in an outpatient setting. Per assignment, each randomized patient had to take two tablets of the investigational product (elobixibat 5 mg or placebo) once a day orally. In the second week, down titration of dose to one tablet or up titration to three tablets was permitted in individual patients based on symptoms. Placebo tablets matching test products in appearance were used.
Allocation concealment
Study drugs (elobixibat or placebo with identical appearance) were supplied in over labeled blister packs and each blister was labeled with a unique kit number. Group assignment was concealed from patients, investigators, sponsors and data analysts.
Implementation of blinding
The patients and investigator (and other personnel involved in the study) were unaware of the study drug(s) administered to patients. The sponsor was also blinded during the study. The placebo tablets and its packaging and labeling were identical in appearance to that of test products.
Study objectives and outcomes
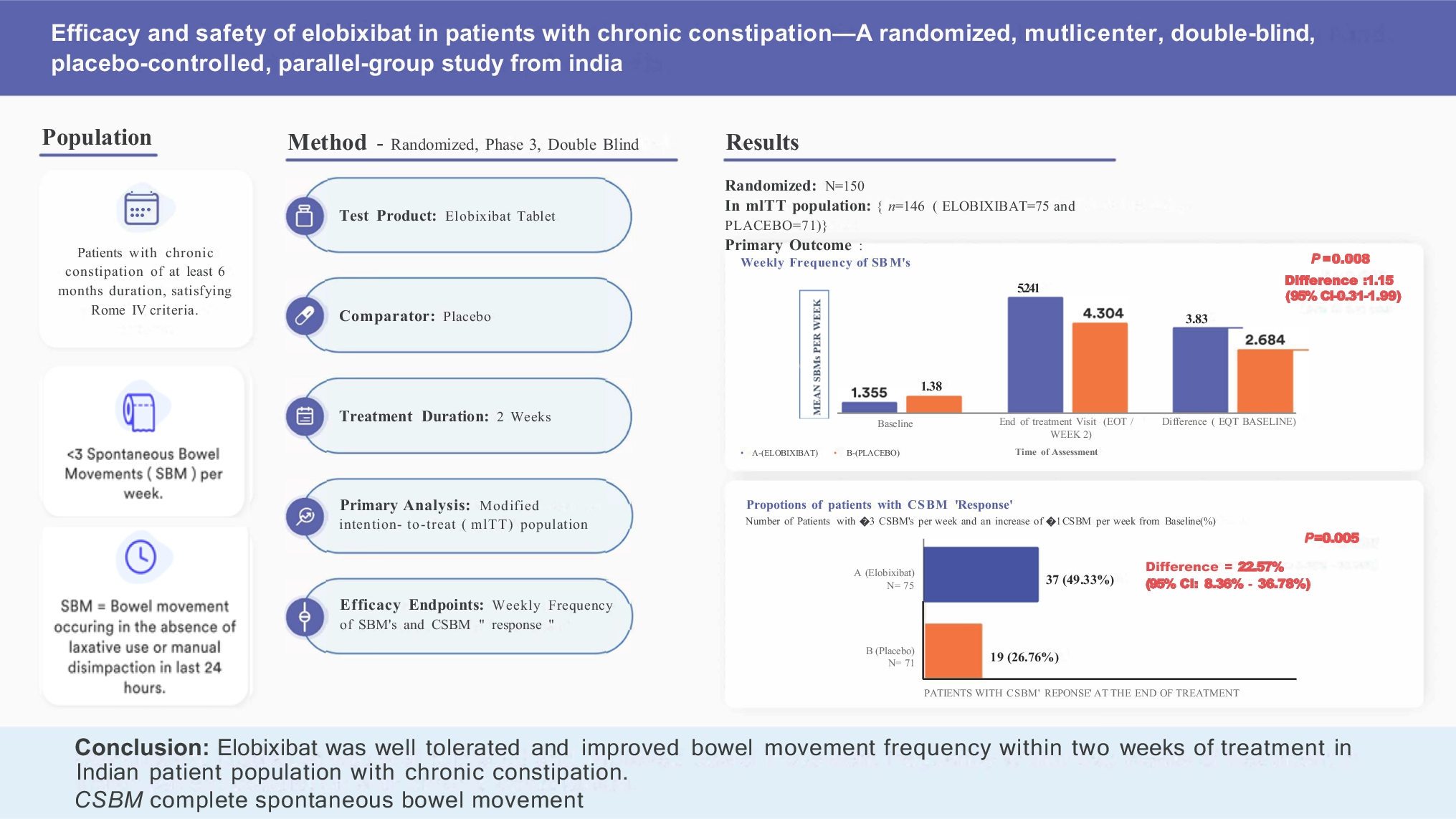
The primary objective of the study was to evaluate the efficacy of elobixibat. Primary efficacy variable was weekly frequency of SBMs. The change in weekly frequency of SBMs at the end of treatment (week two) over baseline was the primary efficacy endpoint in this trial. Each patient’s weekly SBM frequency was calculated using the following formula: weekly SBM frequency = 7 × (total number of SBMs during the treatment period)/(number of days of treatment period considered). Use of rescue medication (5 mg bisacodyl tablets or 10 mg bisacodyl suppositories) was permitted if the patient had no bowel movement for 72 hours or longer and this was recorded in the diary for consideration during the efficacy analysis. Bowel movements that followed within 24 hours of rescue medication usage were excluded from computations of SBM frequency.
Secondary efficacy endpoints included proportion of patients with complete spontaneous bowel movement (CSBM) “response” (CSBM responder was defined as a patient with ≥ 3 CSBMs per week and an increase of ≥ 1 CSBM per week from baseline), proportion of patients with a SBM within 24 hours after the first dose of study drug, median time to first SBM, changes in stool consistency score (scored using the seven-point Bristol Stool Form Scale [BSFS]) [20], degree of straining of SBMs and abdominal bloating score (using the five-point ordinal scale) over baseline.
Determination of sample size
Assuming a significance level of 2.5% (two sided), power of 95%, test group mean response 6, placebo group mean response 2 and a pooled standard deviation (SD) of 6 [17], it was estimated that 60 completed patients per treatment group (a total of 120 patients in the study) would be required to prove superiority of elobixibat. Assuming ~ 20% drop-out rates, 150 patients were planned to be randomized in this study with 1:1 ratio (75 patients per arm) to elobixibat and placebo.
Statistical analysis
All descriptive and inferential statistical analyses were performed using SAS® version 9.4 in a secure and validated environment, unless otherwise noted. Statistical significance was concluded when the p-value was less than 0.05. Continuous data was summarized in terms of the mean, SD, median, 95% CI, minimum, maximum and number of observations, unless otherwise stated. Categorical data were summarized in terms of the number of patients providing data at the relevant time point (n), frequency counts and percentages.
All randomized patients who met all inclusion/exclusion criteria administered at least one dose of assigned investigational product and had at least one post-baseline evaluation of the primary estimate were included in the mITT population.
Mean difference between treatments was analyzed using analysis of covariance (ANCOVA) with change from baseline to two weeks as the dependent variable and treatment group and baseline no. of SBMs, center, age and sex as covariates. The two-sided 95% CI and associated p-value were computed for the least square mean (LSM) difference between the elobixibat and placebo drug.
The proportion difference was tested using Chi-square test to assess whether changes in the efficacy parameters are significant in the groups in comparison (elobixibat vs. placebo) of interest. Non-parametric tests were used where appropriate for evaluation of association between different variables. No multiplicity adjustments were made during analysis of the secondary endpoints.
Safety population included all randomized patients who received at least one dose of the investigational drug were included in the safety population. All safety analyses were based upon the safety population.
Data collection and management
During the study, patients were asked to fill in paper diaries daily in the two-week treatment period. The following assessments were to be recorded by patients in the provided paper diaries: time of each bowel movement, stool consistency (scored with the seven-point Bristol Stool Form Scale [BSFS]), sensation of complete bowel emptying and use of rescue medication. Severity of constipation (using the five-point adjectival scale: none, mild, moderate, severe and very severe) was to be recorded once weekly. Patients continued to provide their daily assessments, their weekly assessments and their use of rescue medicine and any other laxatives, suppositories or enemas up to two weeks or end of treatment (EOT).
Electronic case record forms (CRFs) were used to collect information required for data analysis. When the database was declared to be complete and accurate, the database was locked and unblinded.
Ethical considerations
The study was conducted in compliance with the ethical principles that originate in the Declaration of Helsinki and the International Conference on Harmonization (ICH) guidelines for good clinical practice (GCP). The study was approved by institutional ethics committee of each participating centers. Written informed consent was obtained from each patient prior to screening on the approved informed consent form (ICF). Patients received a defined conveyance allowance in the study but were not compensated in other manner. This trial is registered at CTRI (Clinical Trial Registry of India) with the Trial Registration number CTRI/2022/10/046690 (The final protocol for the study is provided as a Supplementary file-appendix 1).


Comments (0)