
Remember me

Kyoto HeLa cells were maintained in high-glucose DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37 °C with 5% CO2. Stable Kyoto HeLa BAC cell lines expressing proteins with a C-terminal fluorescent protein were generated using BAC recombineering67. This yields near-endogenous expression levels of the fusion protein12,68. In these lines, GFP is part of a modified localization and affinity purification (LAP) tag69, providing a short linker. Stable expression was kept under G418 selection (400 μg ml–1; Gibco). The following BAC lines were used: FUS (MCB_005340; also used for the compound screen), COIL (MCB_0002582), DCP1A (MCB_0003876), EWSR1 (MCB_0008863), PABPC1 (MCB_0006901), TIAL1 (MCB_0008967) and TRP53BP1 (MCB_0003740). HeLa FUS-P525L–GFP cells were generated using the same approach as described previously for the iPS cell lines44.
Human iPS cells were grown in either TeSR E8 or mTeSR1 medium (Stem Cell Technologies) at 37 °C with 5% CO2 (ref. 70). iPS cells lines, derived from two different donors, expressing FUS with a C-terminal GFP fluorescent marker were used. All were generated using CRISPR–Cas9-assisted tagging and mutagenesis and have been previously described44. KOLF iPS cell lines expressing WT FUS–GFP or FUS-P525L–GFP were previously generated from the KOLF-C1 clonal iPS cell line produced as part of the Human Induced Pluripotent Stem Cell Initiative71. KOLF-C1 cells were derived from a healthy male donor. In these lines, GFP is part of a modified LAP tag69, yielding an identical fusion protein sequence to the Kyoto HeLa BAC cell line. AH-ALS1-F58 iPS cells expressing FUS-P525L with a C-terminal GFP fluorescent marker were previously generated from a clonal iPS cell line from a female individual with ALS expressing FUS-P521C. The P525L mutation and GFP tag were introduced and the P521C mutation was corrected by simultaneous tagging and mutagenesis44,72,73.
MNs used for the study of FUS-P525L dynamics were induced and maintained as described previously74. MNs used for the prolonged arsenite stress assay were derived from commercially available WTC-11 iPS cells (Coriell Institute, GM25256) and differentiated as described previously75. MNs used for axonal lysosome mobility assays were generated from AH-ALS1-F58 iPS cells expressing FUS-P525L. In short, the iPS cells were differentiated into neuronal progenitor cells and matured to spinal MNs in Matrigel-coated plates, as previously described44,70. The coating and assembly of silicone microfluidic chambers (MFCs; RD900, Xona Microfluidics) to prepare for subsequent seeding of MNs was performed as described previously44,76,77. MNs were eventually seeded into one side of an MFC for maturation to obtain a fully compartmentalized culture with proximally clustered somata, and their dendrites were physically separated from their distal axons, as only the latter type of neurite was able to grow from the proximal seeding site through a microgroove barrier of 900-µm-long microchannels to the distal site (Fig. 6b). All subsequent imaging in MFCs was performed at day 21 of axonal growth and MN maturation (day 0 was the day of seeding into MFCs).
All procedures using human cell samples were performed in accordance with the Helsinki convention and approved by the Ethical Committee of the Technische Universität Dresden (EK45022009, EK393122012).
Recombinant protein purificationRecombinant proteins were purified using baculovirus/an insect cell expression system, as previously described12. Briefly, 6×His-MBP-FUS–GFP and 6×His-MBP-FUS–SNAP were purified from Sf9 cell lysates by Ni-NTA (Qiagen) affinity purification. The 6×His-MBP tag was cleaved by 3C protease, concentrated by dialysis and further purified by size-exclusion chromatography. 6×His-MBP-SFPQ–GFP was purified from Sf9 cell lysates by affinity purification using Ni-NTA and amylose resin (New England Biolabs). The 6×His-MBP tag and, if necessary, the GFP tag were cleaved by 3C protease and TEV protease, respectively, and the target proteins were concentrated by dialysis and further purified by cation exchange chromatography. The composition of the storage buffer for the purified proteins was 1 M or 500 mM KCl, 50 mM Tris-HCl (pH 7.4), 5% glycerol and 1 mM DTT, and the concentration of FUS was adjusted to 30 μM in storage buffer before use.
Small-molecule screenFor the small-molecule screen, we used the Pharmakon 1600 library of small molecules (MicroSource Discovery Systems) prepared as 10 mM solutions in DMSO. The Kyoto HeLa BAC cell line stably expressing FUS–GFP was seeded at 4,000 cells per well in 384-well plates 24 h before the assay. The cells were pretreated with 10 μM compound for 1 h and stressed with 1 mM potassium arsenate (A6631, Sigma-Aldrich). After 1 h, cells were fixed in 4% formaldehyde and stained with 1 µg ml–1 Hoechst 33342 and CellMask blue (1:10,000; H32720, Thermo Fisher Scientific) before being imaged on a CellVoyager CV7000 automated spinning disc confocal microscope (Yokogawa) with a ×40/1.1-NA air objective to assess FUS–GFP localization.
FUS–GFP signal was analyzed using CellProfiler78, and the data were processed with KNIME. The cytoplasm and nuclei were distinguished with weak (CellMask blue) and strong (Hoechst 33342) blue fluorescent signals, respectively. Particle number and sum area, granularity (at 9, 10 and 11 pixels in the cytoplasm or 1, 5, 6, 7, 8 and 9 pixels in the nucleus) scale, texture (at 10-pixel scale) and integrated signal intensity of FUS–GFP in the nucleus and cytoplasm were measured. Z scores (z = (x – μ) / σ, where x is the observed value, μ is the control mean, and σ is the control standard deviation) relative to the DMSO-treated control wells on each plate were calculated for these parameters and combined into the Mahalanobis distance. Compounds of interest were selected on the criteria of (1) treatment returned the cells to the unstressed state (that is, reduced stress granule number and increased nuclear signal), (2) a clear monotonic dose-dependent response and (3) manual prioritization by known mechanism (for example, emetine and cardiac glycosides) or implausibility as a cell-compatible compound (for example, surfactants used as topical antiseptics).
The follow-up in vitro assay of compounds on FUS–GFP condensates was assessed in a 384-well plate format. The compound volumes (in DMSO) necessary for 1, 3, 10, 30 or 100 μM final concentration were added by acoustic dispensing (Labcyte Echo 550) to 96-well plate wells containing FUS–GFP in 3 µl of 50 mM Tris-HCl (pH 7.4), 1 mM DTT and 170 mM KCl. The final DMSO concentration was 0.01 to 1%. Using a Freedom Evo 200 liquid handling workstation (TECAN), the FUS–GFP/compound mixture was diluted in 7 μl of 50 mM Tris-HCl (pH 7.4) to reach the final composition of 50 mM Tris-HCl (pH 7.4), 1 mM DTT, 50 mM KCl, indicated concentrations of each compound and DMSO and 0.7 μM FUS–GFP. Compound/FUS–GFP and assay buffer were mixed using a standardized pipetting procedure, split into four wells in clear-bottom 384-well plates and immediately imaged using a CellVoyager CV7000 automated spinning disc confocal microscope (as described above). Condensates in suspension for six fields of view were imaged as a maximum intensity projection of six focal planes at 2-μm steps per sample. Condensate number and FUS–GFP partitioned into condensates were analyzed with a fixed intensity threshold using Fiji. The number of condensates and partitioning were weakly time dependent due to condensate sedimentation and therefore normalized assuming a linear change over time by reference to DMSO controls at the start and end of each plate row.
Compound characterization in cellsCompound effects were assessed under a variety of conditions in HeLa cells, iPS cells or iPS cell-derived MNs. Unless otherwise indicated, cells were pretreated for 1 h using 10 μM compound from 10 mM stock in DMSO (or an equal volume of DMSO control) and stressed for 1 h with 1 mM potassium arsenate in the presence of the compounds. Live cells were imaged by widefield epifluorescence using an inverted Olympus IX71 microscope with a ×100/1.4-NA Plan Apo oil immersion objective (Olympus) and a CoolSNAP HQ CCD camera (Photometrics) using a DeltaVision climate control unit (37 °C, 5% CO2; Applied Precision).
Various cellular stresses were achieved by replacing 1 h of potassium arsenate treatment with other conditions: 0.4 M sorbitol (S1876, Sigma-Aldrich) from a 4 M stock in water for 1 h (osmotic stress), 42 °C in normal growth medium for 30 min (heat stress) or 100 mM 6-deoxyglucose (D9761, Sigma-Aldrich) from a 1 M stock in water in glucose-free DMEM (11966025, Thermo Fisher Scientific) supplemented with 10% fetal calf serum for 1 h (glycolysis stress). Sodium arsenite (S7400, Sigma-Aldrich) was used from a 10 mM stock in water.
Other antioxidants were used by replacing lipoamide treatment: l-ascorbic acid (A4544, Sigma-Aldrich) from a 1 M stock in water, N-acetyl l-cysteine (017–05131, Wako) from a 100 mM stock in water, citric acid (036-05522, Wako) from a 1 M stock in water ±-α-tocopherol (209-01791, Wako) from a 100 mM stock in ethanol and taurine (201-00112, Wako) from a 100 mM stock in water.
Compound dose responsesTo assess dose-dependent effects of lipoamide on HeLa and iPS cells expressing FUS–GFP, cells were pretreated with lipoamide for 1 h followed by 1 h of treatment with 1 mM potassium arsenate, similar to the ex vivo HeLa cell screen except serial compound dilutions in medium were prepared manually from 80 μM to ~0.4 nM at 1.189× dilution steps. Small dilution steps rather than concentration replicates were selected as this approach provides greater statistical power from a set number of samples79. The final DMSO concentration was 0.08% in all samples, and each plate included at least 12 control wells with 0.08% DMSO. Cytoplasmic FUS–GFP condensate number and nuclear/cytoplasm partitioning of FUS–GFP were analyzed using custom macros in Fiji. Nuclei were identified by intensity thresholding of DNA images labeled with Hoechst following a 5-pixel Gaussian blur. Cytoplasmic FUS–GFP condensates were identified by intensity thresholding of FUS–GFP images following a 10-pixel weight 0.9 unsharp filter masked by the thresholded nuclei. The ratio of the number of cytoplasmic FUS–GFP condensates to that of nuclei was taken as cytoplasmic FUS–GFP condensates per cell per field of view, and p, the ratio of partitioning of FUS–GFP to the nucleus and the cytoplasm, was derived from a = vn / vt, the ratio of nuclear to total green signal per field of view, where p = a / (1 – a). These data were log transformed and fitted to a Rodbard sigmoidal curve80 to determine EC50. Six fields of view were captured and analyzed per condition.
The series of lipoamide analogs including lipoamide and lipoic acid were newly synthesized by Wuxi AppTec and provided through Dewpoint Therapeutics. To assess the dose–response effects, HeLa BAC cells expressing FUS–GFP were seeded in 384-well plates (4,000 cell per well) 24 h before treatment, pretreated with the compounds in a half-log dilution series (from 30 µM to 3 nM: seven concentrations) using an Echo 650 and treated for 1 h with 1.5 mM potassium arsenate before fixation with 4% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min and counterstained with Hoechst and CellMask blue as described above. Imaging was performed using an Opera Phenix (PerkinElmer; ×20, nine fields of view, binning 2) and Harmony 4.9 software to determine cytoplasmic FUS–GFP condensate number and cytoplasmic and nuclear FUS–GFP intensities to calculate nuclear-to-cytoplasmic ratios of FUS–GFP intensities. EC50 was calculated using either CDD Vault curve fitting or Harmony 4.9 software.
In vitro protein condensation, solidification and oxidationFor the condensation assay at different KCl concentrations, FUS–GFP proteins in storage buffer were diluted with 20 mM HEPES (pH 7.25) containing DMSO and lipoamide to yield 20 µl of the indicated concentrations of the protein and KCl, 0.3 mM DTT and 300 µM lipoamide (0.3% DMSO) and placed on a 384-well plate (781096, Greiner). Condensates were imaged on a Nikon TiE inverted microscope with a Nikon Apo ×60/1.2-NA water immersion objective using a Yokogawa CSU-X1 spinning disk head and an Andor iXon EM + DU-897 EMCCD camera
The assay to determine dilute-phase concentrations at different temperatures was performed with a newly established technique, which will be reported in detail elsewhere. In brief, the technique is based on mass and volume conservation and defined reaction volumes. We can use this method to determine accurate values for both dilute and condensed branch protein concentrations. Here, FUS–GFP phase separation was induced for a protein concentration titration in water-in-oil emulsions in a buffer containing 25 mM Tris-HCl (pH 7.4), 150 mM KCl, 1 mM DTT and the indicated concentrations of lipoamide (or DMSO as control) and imaged with a CSU-W1 (Yokogawa) spinning disk confocal system on an IX83 microscope with a UPlanSApo ×40/0.95-NA air objective, controlled via CellSens (Olympus). The dilute-phase protein concentration was derived from a linear fit to the volume of fractions of condensed-phase FUS–GFP versus the total concentrations of FUS–GFP. Temperature was controlled using a custom-made stage81.
For solidification assays, FUS–GFP in storage buffer was diluted in 50 mM Tris-HCl (pH 7.4) and 1 mM DTT to yield 10 μM protein, 50 mM Tris-HCl (pH 7.4), 1 mM DTT and 50 mM KCl in a volume of 20 μl in nonbinding, clear-bottom 384-well plates (781906, Greiner). Compounds (or an equal volume of DMSO) were then added for a final compound concentration of 30 μM and 0.3% DMSO. ‘Aging’ to cause fiber formation was induced by horizontal shaking at 800 rpm at room temperature, as previously described12. Fiber and condensate formation were analyzed by using a widefield DeltaVision Elite microscope (GE Healthcare Life Sciences) with a Plan ApoN ×60/1.4-NA oil immersion objective (Olympus) and an sCMOS camera (PCO). FRAP of FUS–GFP condensates and fibers was performed on a Nikon TiE inverted microscope with a Nikon Apo ×100/1.49-NA oil immersion objective using a Yokogawa CSU-X1 spinning disc head and an Andor iXon EM + DU-897 EMCCD camera. Regions (10 × 10 pixels) were bleached for 50 ns with a 6-mW, 405-nm laser using an Andor FRAPPA beam delivery unit and imaged for 5 min at 5 Hz. Recovery curves were derived using scripts in Fiji.
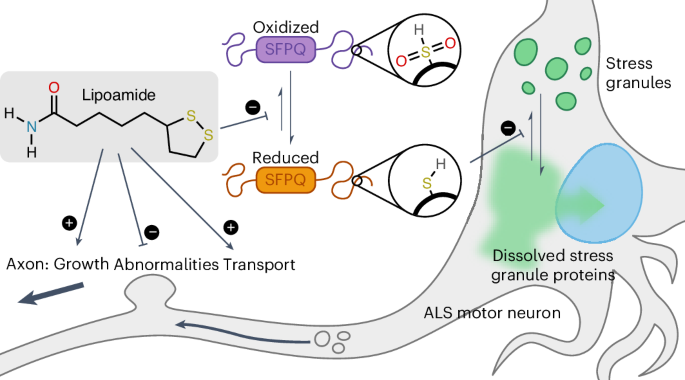
Oxidation of SFPQ was detected by change in mobility in SDS–PAGE without reducing agents. Untagged SFPQ (10 µM) in buffer (20 mM HEPES (pH 7.25) and 150 mM KCl) was incubated with H2O2 at room temperature for 30 min before subjecting to SDS–PAGE. For condensation assays of individual proteins with H2O2, SFPQ–GFP and FUS–GFP condensates were induced in buffer (20 mM HEPES (pH 7.25) and 75 mM KCl). Assays for dissolution and revival of FUS–SNAP condensates were performed in buffer (20 mM HEPES (pH 7.25) and 150 mM KCl). FUS–SNAP was labeled with SNAP-Surface Alexa Fluor 546 (New England Biolabs), and protein mixtures were oxidized with H2O2 at room temperature for 1 h before image acquisition. Proteins were imaged similar to the FUS–GFP condensates described above.
Controlled droplet fusion using optical tweezersLiquidity of FUS protein condensates was assessed by controlled fusion experiments using dual-trap optical tweezers, as detailed previously12,37. In short, for each independent fusion event, two FUS protein droplets in the presence of 300 µM lipoamide or an equivalent amount of DMSO (0.3%) as the control were trapped in each optical trap and brought into contact to initiate droplet coalescence. Fusion relaxation times were accurately recorded as changes to the laser signal as condensate material flowed into the space between the two optical traps during coalescence. The laser signal was recorded at 1 kHz, smoothed at 100 Hz and used to extract the characteristic relaxation time. After fusion was complete (as indicated by a stable laser signal), the fused droplet was discarded, and two new droplets were captured for quantifying an independent fusion event.
Ex vivo DNA cut assaysUV microirradiation was performed as previously described12,44. Briefly, iPS cells expressing WT FUS–GFP were stressed by the addition of 1 mM arsenate for 1 h and treated with lipoamide or an equal volume of DMSO for 1 h. A single point in the nucleus was subject to three UV pulses as described for FRAP but at 10% laser power. GFP fluorescence was imaged at 1 Hz, and the intensity of the response was analyzed on Fiji. iPS cell MNs expressing FUS-P525L–GFP were pretreated with 20 µM lipoamide for 24 h before laser irradiation. The UV laser cutter setup used a 355-nm UV-A laser with a pulse length of <350 ps. A Zeiss α Plan-Fluar ×100/1.45-NA oil immersion objective was used, and 12 laser shots in 0.5-µm steps were administered over a 12-µm linear cut.
Nuclear magnetic resonance for FUS–lipoamide interactionUntagged FUS low-complexity domain (residues 1 to 163) was expressed, purified and analyzed using 1H-15N heteronuclear single quantum coherence NMR and sample conditions as previously described82 in the presence of 500 μM lipoamide or equivalent DMSO solvent control (1%).
[15N]-Labeled lipoamide nuclear magnetic resonance[15N]-Racemic (±) and (R)-(+)-lipoamide were synthesized from racemic and (R)-(+)-lipoic acid, respectively, by activating the carboxylic acid using N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. The NHS derivative was reacted with 15NH4Cl to incorporate the [15N] labeling. Full details of the synthesis and subsequent biophysical validation are included in Supplementary Note 1.
For NMR quantification of [15N]-labeled lipoamide, 1H detected 15N edited 1H sensitivity enhanced HSQC NMR ((15N)1H) spectra were acquired on a 14.1T Varian Inova spectrometer equipped with a 5-mm z-axis gradient triple-resonance room temperature probe. Free induction decay was recorded for an acquisition time of 0.0624 s and spectral width of 8 kHz recorded over 1,000 points and a recovery delay of 1 s. Typically, 10,000 transients were collected, yielding a total experiment time of 3 h and 1 min. J coupling between the amide protons and the 15N in water samples was determined to be 88 Hz, and so the transfer times of 1/4 J in the INEPT portions of the pulse sequence were set to 2.6 ms. With these settings, [15N]ammonia or ammonium ions would not be detectable. Chemical modification of [15N]lipoamide (including covalent attachment to an apoenzyme) would yield a substantial change in the (15N)1H NMR spectrum. Similarly, dissolution of lipoamide in a phospholipid membrane would yield substantial peak broadening in the cell samples. We observed neither, consistent with freely diffusing lipoamide.
For optimization of the NMR measurement conditions of [15N]-labeled lipoamide, solvent, pH and temperature sensitivity of the primary amide proton chemical shifts were determined using dummy samples assembled from the appropriate solvent and added compounds.
Integrated NMR signal intensity is proportional to concentration if provided conditions (temperature and pH) are identical83. Chemical exchange84, expected as the amide protons in lipoamide should be labile in water, must also be accounted for. To select appropriate conditions, we determined temperature (Extended Data Fig. 3f) and pH (Extended Data Fig. 3g) sensitivity of the amide proton signal of 1 mM [15N]lipoamide in cell medium. Both amide protons showed chemical exchange under high-temperature, high-pH conditions, with the trans-amide proton affected weakly (Extended Data Fig. 3f,g). We then assessed degradation of the trans-amide proton over 10 h (Extended Data Fig. 3h). At 37 °C, but not 10 °C, the signal intensity decayed slowly, suggesting slow hydrolysis to form ammonia. We concluded that at 10 °C and below pH 8.6, the integrated signal from the trans-amide proton resonance is a good measure of [15N]lipoamide concentration.
For quantification of [15N]lipoamide cellular uptake, HeLa cells expressing FUS–GFP were grown in six-well plates to 106 cells per well in DMEM supplemented with 10% fetal calf serum. To simultaneously stress and treat cells, the medium was replaced with 0.6 ml of medium supplemented with potassium arsenate and 100 μM [15N]-racemic (±) or (R)-(+)-lipoamide for 1 h at 37 °C. High concentrations of compound were used to maximize the signal. The medium was then removed and retained (medium sample), the cells were washed with ~2 ml of PBS, and the cells were removed by trypsinization with 0.3 ml of TrypLE Express (12604013, Thermo Fisher Scientific) and incubation at 37 °C for 5 min; 0.3 ml of medium was added to quench the trypsin. The resuspended cells were retained (cell sample). All samples were frozen at −80 °C. Wells were prepared for all combinations of no compound (1% DMSO control), [15N](±)-lipoamide or [15N]-(R)-(+)-lipoamide, with or without potassium arsenate and with or without cells.
The concentrations of [15N]lipoamide inside (Ccell) and outside (Cout) the cells were calculated from measurements of signal intensity S of the trans-amide proton of lipoamide acquired in the absence (−cells, sample i; Extended Data Fig. 3a) and presence (+cells, sample ii; Extended Data Fig. 3a) of HeLa cells, using the following equations:
$$_}}}=\left(1-U\;\right)\frac_}}}_}}}}_}}}}$$
$$_}}}=U\frac_}}}_}}}}__}}}}$$
where Ncell = 106, cadd = 100 μM, and Vadd (added volume) = 600 μl. V1 = 4.19 × 10−15 m3 (approximating HeLa cells as spheres of radius 10−5 m), and U represents measured fractional uptake as given by
To measure in vitro partitioning of [15N](±)-lipoamide in the FUS–GFP condensate phase, phase separation of FUS–GFP, at room temperature, was achieved by diluting 12.5 μl of protein stock at 170 μM (in salty HEPES buffer: 50 mM HEPES, 500 mM KCl, 5% glycerol (pH 7.25) and DTT 1 mM) with 247 μl of salt-free buffer containing [15N](±)-lipoamide (50 mM HEPES, 5% deuterium oxide, 105 μM [15N](±)-lipoamide and 1.05% DMSO (pH 8)), resulting in samples of 260 μl with 8 μM FUS–GFP, 100 μM [15N](±)-lipoamide and 25 mM KCl.
The sample was centrifuged for 10 min at 4,000g at room temperature, and the supernatant was kept for NMR analysis. The remaining supernatant was carefully pipetted out, without disturbing the condensate pellet, and discarded. Perpendicular view photographs of the pellet were taken. Finally, the condensate pellet was resuspended in 260 μl of buffer with the same buffer conditions as the phase-separated sample (50 mM HEPES and 25 mM KCl (pH ~7.4)).
Resuspended condensate or supernatant was loaded in deuterium oxide-matched 5-mm Shigemi tubes and analyzed by (15N)1H NMR. To achieve adequate signal to noise, the resuspended condensate was scanned for 20 h, whereas the supernatant was scanned for 4 h. The signal factor (intensity ratio between the dilute and resuspended condensate samples) was adjusted accounting for differences in sample volume and the number of scans.
The volume of condensate was calculated from perpendicular photographs (see Extended Data Fig. 4c) from the pellet radius (a) and inner radius of the semispherical bottom of the microcentrifuge tube (r):
$$V=\frac}}^\left(2+\cos }\right)}\right)}^$$
θ indicates the subtended angle, as showed in Extended Data Fig. 4c.
The ratio between the concentration of lipoamide in the condensate and dilute phases (partition coefficient (PC)) was calculated using
$$=\,\frac_}}}+_}}}}* _}}}}$$
where VCond is the volume of condensate phase, VAdded is the volume added to resuspend the condensate phase (260 μl), and SF is the signal intensity ratio between the dilute phase and the resuspended condensate measured by NMR.
The concentrations of lipoamide in the condensate ([L]Cond) and dilute ([L]Dil) phases were calculated as
$$_}}}=\,\frac_}}}* _}}}}_}}}+\,\frac_}}}-_}}}}}}\,$$
where [L]Tot is the total concentration of [15N](±)-lipoamide, and VTot is the total volume of the phase-separated sample. The fraction (%) of [15N](±)-lipoamide signal in the condensate phase (see Extended Data Fig. 4d) was calculated as
$$}\; }\; }\; }}\left( \% \right)=\frac}* 100.$$
For derivation of these expressions, see ‘Derivation of NMR calculations’.
Cross-linking coupled to mass spectrometryFUS condensates were processed and analyzed essentially as described previously85. In short, reconstituted droplets of lysine-rich FUS-K12 or FUS-G156E were generated by low salt (80 mM KCl) and cross-linked by the addition of H12/D12 DSS (Creative Molecules) in the presence or absence of lipoamide for 30 min at 37 °C with shaking at 600 rpm. Protein samples were quenched by the addition of ammonium bicarbonate to a final concentration of 50 mM and directly evaporated to dryness. The dried protein samples were denatured in 8 M urea, reduced by the addition of 2.5 mM TCEP at 37 °C for 30 min and subsequently alkylated using 5 mM iodoacetamide at room temperature for 30 min in the dark. Samples were digested by the addition of 2% (wt/wt) trypsin (Promega) overnight at 37 °C after adding 50 mM ammonium hydrogen carbonate to a final concentration of 1 M urea. Digested peptides were separated from the solution, retained by a C18 solid-phase extraction system (SepPak Vac 1cc tC18 (50-mg cartridges, Waters)) and eluted in 50% acetonitrile and 0.1% formic acid. Dried peptides were reconstituted in 30% acetonitrile and 0.1% trifluoroacetic acid and separated by size-exclusion chromatography on a Superdex 30 increase 3.2/300 column (GE Life Sciences) to enrich for cross-linked peptides. Peptides were subsequently separated on a PepMap C18 2 µM, 50 µM × 150 mm (Thermo Fisher Scientific) column using a gradient of 5 to 35% acetonitrile for 45 min. Mass spectrometry measurements were performed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) in data-dependent acquisition mode with a cycle time of 3 s. The full scan was performed in the Orbitrap with a resolution of 120,000, a scan range of 400–1,500 m/z, an automatic gain control target of 2.0 × 105 and an injection time of 50 ms. Monoisotopic precursor selection and dynamic exclusion were used for precursor selection. Only precursor charge states of 3–8 were selected for fragmentation by collision-induced dissociation using 35% activation energy. MS2 was performed in the ion trap in normal scan range mode with an automatic gain control target of 1.0 × 104 and injection time of 35 ms. Data were searched using xQuest in ion-tag mode. Carbamidomethylation (+57.021 Da) was used as a static modification for cysteine. Cross-links were quantified relative to the condition containing no lipoamide.
Cultured cell transfectionTransfection for gene depletion was performed with Lipofectamine 2000 (Thermo Fisher Scientific) and esiRNA oligonucleotides targeting human genes (Eupheria Biotech), as listed in Supplementary Table 1. esiRNA targeting Renilla luciferase was used as a negative control. The medium was replaced 5 h after transfection, and the cells were cultured for 3 days before analysis.
Immunocytochemistry of cultured cellsHeLa or U2OS cells were fixed with 4% paraformaldehyde (PFA) in PBS at room temperature for 15 min and washed with PBS containing 30 mM glycine. After permeabilization with 0.1% Triton X-100 in PBS at 4 °C and a following wash with glycine-containing PBS, cells were blocked with 0.2% fish skin gelatin (Sigma) in PBS (blocking buffer) at room temperature for 20 min, incubated with primary antibodies in blocking buffer overnight at 4 °C, washed with blocking buffer and incubated with secondary antibodies and DAPI in blocking buffer at room temperature for 1 h. After washing with blocking buffer, the samples were stored in PBS until imaging. For detection of endogenous SFPQ, cells were fixed with cold methanol on ice for 10 min and blocked with blocking buffer at room temperature for 20 min before treatment with primary antibodies. Samples were imaged on a Zeiss LSM 700 or 880 confocal microscope with a ×40/1.2-NA water objective (Zeiss) or Opera Phenix Plus High-Content Screening System with a ×20/1.0-NA water objective (Revvity). Segmentation of cell nuclei, the cytoplasm, stress granules and mitochondria and measurement of fluorescence intensities at each segment were performed using CellProfiler. The data were then processed using KNIME to calculate the number of stress granules per cell and nuclear-to-cytoplasmic intensity ratios of stress granule proteins. For the click-crosslink lipoamide analog, intensity ratios of the click-cross-link lipoamide analog at stress granules, mitochondria and nuclei to the cytoplasm (excluding stress granules and mitochondria) intensity; inrensity ratios without cross-linking to with cross-linking and percentage of cells that had more than two stress granules.
iPS cell MNs were fixed for 15 min at room temperature in 4% PFA in PBS. Permeabilization and blocking were performed simultaneously using 0.1% Triton X-100, 1% bovine serum albumin (BSA) and 10% FBS in PBS at room temperature for 45 min. Subsequently, primary antibodies were applied overnight at 4 °C in 0.1% BSA in PBS. The cells were washed with 0.1% BSA in PBS and incubated with secondary antibodies for 1 h at room temperature. Finally, the cells were washed three times with 0.1% BSA in PBS supplemented with 0.005% Tween-20, including Hoechst or DAPI in the second washing step. Neurofilament H was used as a marker of MNs. Samples were imaged on either a CellVoyager CV7000 automated spinning disc confocal microscope (Yokogawa) with a ×40/1.3-NA water objective or a Zeiss LSM880 laser scanning confocal microscope.
The following primary antibodies were used: rabbit anti-G3BP1 (PA5-29455, Thermo Fisher Scientific), mouse anti-TOM20 (F-10, Santa Cruz), mouse anti-SPFQ (C23, MBL), mouse anti-SRSF1 (103, Invitrogen), rabbit anti-TDP-43 (80002-1-RR, Proteintech), mouse anti-SC35 (ab11826, Abcam), rabbit anti-SP100 (HPA016707, Sigma-Aldrich), mouse anti-NPM1 (FC82291, Sigma-Aldrich), rabbit anti-HP1α (2616S, Cell Signaling), mouse anti-neurofilament H (SMI-32, Millipore) and mouse anti-β3-tubulin (T5076, Sigma-Aldrich). The following secondary antibodies were used: Alexa Fluor 488-conjugated anti-mouse, Alexa Fluor 594-conjugated anti-rabbit and anti-mouse and Alexa Fluor 647-conjugated anti-rabbit and anti-mouse (Thermo Fisher Scientific).
UV cross-linking and click reactionHeLa cells were treated with 3 mM arsenate for 1 h, followed by 30 µM of the click-cross-link lipoamide analog for 30 min in the presence of arsenate. The cells were then irradiated with a 305-nm light-emitting diode for 10 s for cross-linking just before fixation with 4% PFA in PBS at room temperature. The fixed cells were subjected to immunostaining as described above. After staining, the cells were subjected to click reaction with 2 µM AF594-Picolyl-Azide (CLK-1296-1, Jena Bioscience) in buffer containing 100 mM HEPES (pH 7.25), 5 mM l-ascorbic acid, 0.5 mM THPTA and 0.1 mM CuSO4 at 37 °C for 40 min. Cells were then washed three times with 0.1% Triton X-100 in PBS to remove free dye. Imaging was performed on a CSU-W1 (Yokogawa) spinning disk confocal system on an IX83 microscope (Olympus) with a UPlanSApo ×100/1.4-NA oil objective (Olympus).
Treatment with l-azidohomoalanineWT HeLa cells were first washed with and cultured in methionine-free medium (21013-24, Gibco) supplemented with 10% FBS for 1 h. The medium was then replaced with complete medium or methionine-free medium supplemented with 1 mM methionine (M9625, Sigma-Aldrich) or AHA (C10102, Invitrogen) for 2 h before the cells were stressed with 1 mM arsenate for 1 h. After fixation with 4% PFA in PBS, the cells were subjected to immunostaining to label G3BP1.
ImmunoblottingFor the puromycin incorporation assay, HeLa cells were treated with 10 µM lipoamide or control 0.1% DMSO for 1 h, followed by 1 mM arsenate for an additional 1 h in the presence of lipoamide. The cells were then treated with 91.8 µM (50 µg ml–1) puromycin (Sigma, P8833) for 5 min before being washed with PBS and lysed with a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1% SDS, protease inhibitors and PhosSTOP (Roche). For testing SFPQ protein levels, HeLa cells treated with AHA as described above were washed with PBS and lysed with 1% NP-40 and 0.1% SDS in PBS on ice for 15 min. The lysates were clarified by centrifugation at 20,000g for 10 min, separated by SDS–PAGE, transferred onto nitrocellulose membranes and subjected to immunoblotting. The following primary antibodies were used: rabbit anti-SFPQ (ARP4-572, Aviva Systems Biology), mouse anti-α-tubulin (DM1a, Sigma-Aldrich), mouse anti-puromycin (12D10, Merk Millipore) and mouse anti-GAPDH (G8795, Sigma). IRDye 800CW and IRDye680RD (LI-COR) were used as secondary antibodies to detect signal using the Odyssey platform (LI-COR).
Time-lapse cellular imagingTime-lapse imaging was performed at 37 °C with 5% CO2. iPS cell-derived MNs were treated with 20 µM lipoamide or 0.02% DMSO (control) for 1 h and then treated with 20 µM arsenite just before image acquisition. Maximum projection images were generated, and the number of FUS–GFP+ foci was quantified by Fiji.
Axonal transport assaysAH-ALS1-F58 iPS cell MNs expressing FUS-P525L were treated with 2 μM compound or an equal volume of DMSO for 3 days. Longer incubation was selected to ensure penetration and action of compounds along the length of the axon channel. A concentration of 2 μM was selected as the highest concentration where there were no toxic effects on this iPS cell line (assessed qualitatively). Analysis of axonal transport of lysosomes was performed as previously described44. Briefly, lysosomes were labeled by the addition of 50 nM lysotracker red (Thermo Fisher Scientific) and imaged using a Leica DMI6000 inverted microscope with a ×100/1.46-NA oil immersion objective and an Andor iXON 897 EMCCD camera in an incubator chamber (37 °C, 5% CO2) at 3 Hz for 120 s at either the proximal or distal end of the silicone channels harboring the axons. Kymographs were generated using Fiji. Particle tracking was used to identify proportion of particles moving faster than 0.2 μm s–1 for five videomicrographs. Each video includes a variable population of nonmoving background particles; therefore, for each biological replicate, data were normalized to the mean proportion of moving lysosomes (>0.2 µm s–1) at either MFC site (proximal and distal) in the DMSO (solvent control)-treated FUS-P525L samples in Fig. 6d.
Protein aggregation in C. elegansThe effect of lipoic acid on stress granule protein aggregation in vivo was analyzed using a C. elegans model for stress granule formation and aggregation. As previously described, fluorescent-tagged PAB-1 forms abundant stress granules and large solid aggregates during aging or following chronic stress47,48. RHO-1 also aggregates during aging but is not an RNA-binding or stress granule protein. Two lines were used: fluorescently tagged PAB-1 (DCD214: N2; uqIs24[pmyo-2::tagrfp::pab1gene]) and RHO-1 (DCD13: N2; uqIs9[pmyo-2::rho-1::tagrfp+ptph-1::gfp]). Each line was analyzed as described below, except DCD13, which was maintained at 20 °C.
Animals were exposed to lipoic acid in liquid culture in a 96-well plate starting from larval stage 4 (L4) in a total volume of 50 μl of S-Complete per well (100 mM NaCl, 50 mM potassium phosphate (pH 6), 10 mM potassium citrate, 3 mM MgSO4, 3 mM CaCl2, 5 μg ml–1 cholesterol, 50 μM EDTA, 25 μM FeSO4, 10 μM MnCl2, 10 μM ZnSO4 and 1 μM CuSO4) supplemented with heat-killed OP50 and 50 μg ml–1 carbenicillin. Per experiment, a minimum of nine wells each with 13 animals were treated with (R)-(+)- or (S)-(–)-lipoic acid or an equivalent volume of DMSO.
Forty-eight hours after switching L4 animals from 20 °C to 25 °C (day 2 of adulthood), extensive aggregation of fluorescently tagged PAB-1 and RHO-1 occurs in the pharyngeal muscles. After immobilization with 2 mM levamisole, aggregation was scored using a fluorescence stereo microscope (Leica M165 FC, Plan Apo ×2.0 objective). For PAB-1, aggregates occurred primarily in the terminal bulb of the pharynx, and aggregation was scored by the number of aggregates (more than ten per animal). For RHO-1, aggregates were scored in the isthmus of the pharynx, and aggregation was scored as high (>50% of the isthmus), medium (<50%) or low (no aggregation). High-magnification images were acquired with a Leica SP8 confocal microscope with an HC Plan Apo CS2 ×63/1.40-NA oil objective using a Leica HyD hybrid detector. tagRFP::PAB-1 was detected using 555 nm as excitation and an emission range from 565 to 650 nm. Representative confocal images are displayed as maximum z-stack projections.
D. melanogaster ALS modelsAll fly stocks were maintained on standard cornmeal at 25 °C in a light-/dark-controlled incubator. w1118, UAS-eGFP, D42-GAL4 and OK6-Gal4 flies were obtained from Bloomington Drosophila Stock Center. UAS-FUSWT, UAS-FUSP525L and UAS-FUSR521C flies were previously described49,86. UAS-TDP-43WT and UAS-TDP-43M337V flies were provided by J. P. Taylor (St. Jude Children’s Research Hospital, Memphis, Tennessee, USA)87.
Tissue-specific expression of human genes was performed with the Gal4/UAS system88. Climbing assays were performed as previously described86. Briefly, flies expressing eGFP, human FUS or TDP-43 were grown in the presence or absence of lipoic acid (430 µM; ethanol was used as the vehicle control) or lipoamide (430 µM; DMSO was used as the vehicle control) and anesthetized, placed into vials and allowed to acclimate for 15 min in new vials. Feeding these compounds did not show obvious lethality or toxicity at these concentrations. For each fly genotype, the vial was knocked three times on the base on a bench, and the number of flies climbing up the vial walls was counted. The percentage of flies that climbed 4 cm in 30 s was recorded. TDP-43-expressing flies were raised at 18 °C to suppress lethality.
For immunohistochemistry of neuromuscular junctions, parent flies were crossed on food supplemented with DMSO or lipoamide, and offspring were raised on the same food. Wandering third instar larvae were dissected and subjected to immunostaining as described previously89. Briefly, the dissected larvae were fixed with 4% PFA in PBS at room temperature for 20 min and washed with PBS. After removing unnecessary tissues, the samples were blocked with 0.2% fish skin gelatin (Sigma) and 0.1% Triton X-100 in PBS (blocking buffer) at room temperature for 1 h, incubated with anti-HRP-Cy3 (1:200; Jackson Immunoresearch) in blocking buffer overnight at 4 °C, washed with 0.2% Triton X-100 in PBS and incubated with Alexa Fluor 488 Phalloidin (1:5,000; Thermo Fisher Scientific) at room temperature for 2 h to visualize muscles. The samples were then washed with 0.2% Triton X-100 in PBS and mounted with 70% glycerol in PBS. Synaptic boutons of muscle 4 in abdominal segments 2, 3 and 4 (A2–A4) were imaged using a Zeiss LSM 700 or 880 confocal microscope with a ×40/1.2-NA water objective (Zeiss). Numbers of synaptic boutons and satellite boutons were counted manually.
Quantitative PCR with reverse transcriptionQuantitative PCR was performed using primer sequences targeting GAPDH (control) and full-length STMN2 exactly as previously described90.
Thermal proteome profilingTPP was performed as described previously34. Briefly, two 150-mm dishes of HeLa cells (~6 million cells per dish) were treated with 0.1% (vol/vol) DMSO (control) or 100 µM lipoamide for 1 h. At the end of incubation, one lipoamide-treated dish and one DMSO-treated dish were stressed with 1 mM arsenate for 1 h. The second set of cells served as the control (treatment with water, vehicle in which arsenate was dissolved) for only lipoamide treatment and only DMSO treatment. All incubations were performed at 37 °C with 5% CO2. Following incubation, cells were washed with PBS and trypsinized. Cells were collected by centrifugation at 300g for 3 min. The cell pellet was resuspended in PBS containing the appropriate treatment concentrations of the compounds (lipoamide, DMSO and arsenate) at a cell density of 4
Comments (0)