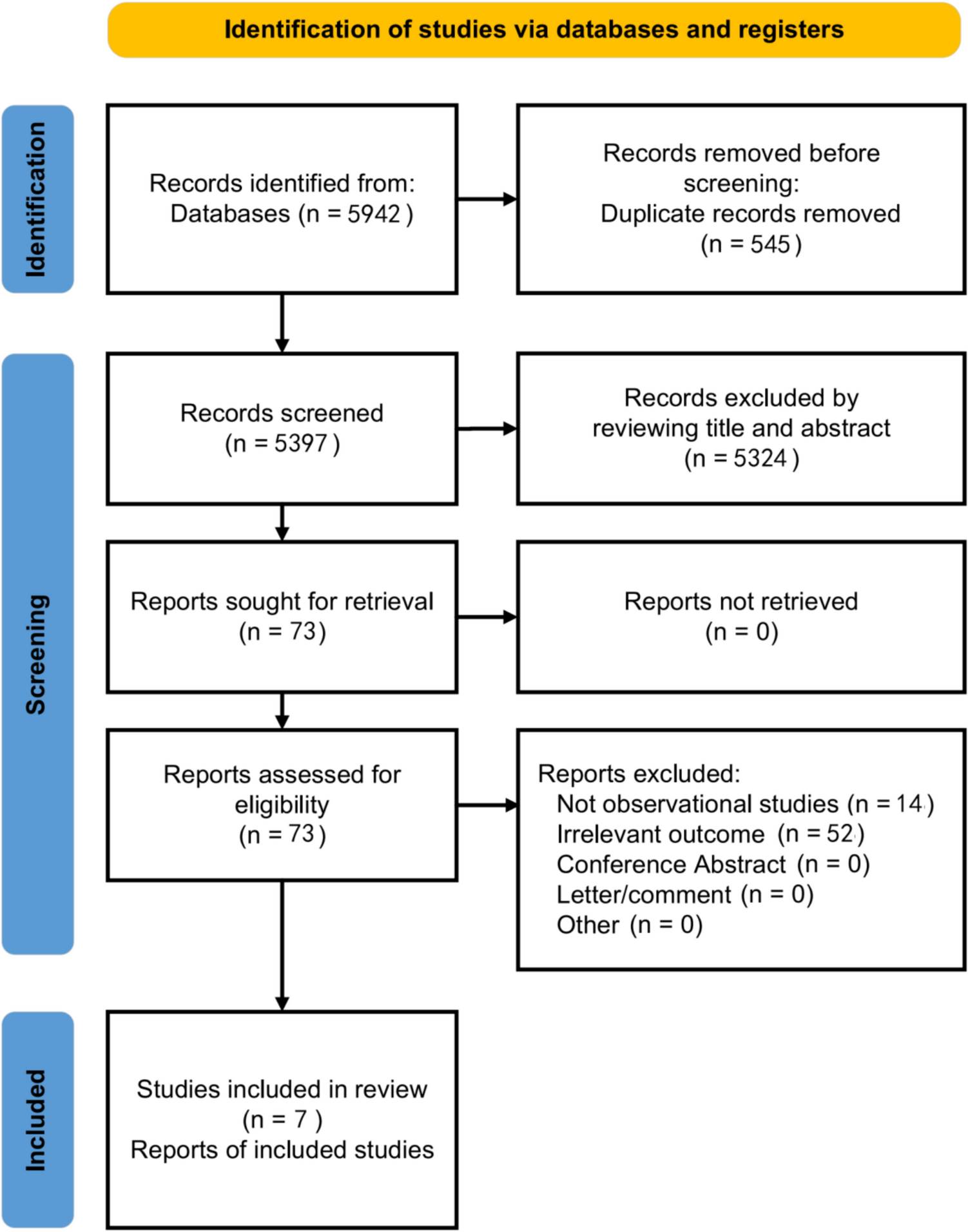
Dataset and pre-processing
A TCGA-LUSC dataset (496 LUSC samples and 49 normal samples), as well as associated survival information, was provided by the UCSC Xena database [15]. Meanwhile, the dataset GSE73403 and associated survival information in Gene Expression Omnibus (GEO) database [16] were enrolled in this investigation. GSE73403 contained 69 samples from LUSC patients. The data were generated on the platform of GPL6480 Agilent-014850. Using the probe expression matrix and the annotation file, we excluded probes that did not match any gene symbols. When multiple probes were linked to a single gene, the average expression value of those probes was calculated for that gene. Furthermore, we identified 277 genes associated with PANoptosis from a previously published study by Song et al. [17].
PANoptosis-related DEGs investigation
DEGs between LUSC and normal samples from TCGA-LUSC were analyzed using the DESeq2 package in R [18]. In summary, the significance of gene expression was assessed based on log2 fold change (FC) > 1 and P value < 0.05. The results were then visualized with a volcano plot. Then, a VENN plot analysis was performed based on DEGs and PANoptosis-associated genes to explore the PANoptosis-related DEGs by using ggvenn package in R (version: 0.1.10).
Functional enrichment and protein–protein interaction (PPI) network analysis
GO function and KEGG pathway analyses were conducted on PANoptosis-related DEGs using clusterProfiler package of R [19] with the cutoff value of P value < 0.05. Then, according to STING database (version: 11.0) [20], the protein interaction information was extracted, and PPI pairs (median confidence = 0.4) among PANoptosis-related DEGs were predicted. Finally, the Cytoscape (version: 3.7.1) software [21] was applied to visualize the current network.
Clustering analysis for PANoptosis-related DEGs
To investigate different clusters of LUSC associated with PANoptosis, the ConsensusClusterPlus package [22] in R software was used for analysis based on TCGA-LUSC dataset. Kaplan–Meier (KM) survival analysis was carried out on clusters based on the Survival package (version 2.41-1). Subsequently, the ESTIMATE algorithm [23] was then utilized to investigate the stromal scores, immune scores, ESTIMATE scores, and tumor purity across various clusters. A P value below 0.05 was deemed statistically significant. The findings were represented using a box plot.
Prognostic model construction and validation
The univariate Cox regression investigation was applied on PANoptosis-related DEGs to reveal prognostic genes with P < 0.05. Using LASSO Cox regression in R [24], the optimal gene set was identified from prognostic genes in the TCGA-LUSC training dataset through tenfold cross-validation with the glmnet package. Then, based on the prognostic coefficient of key genes obtained above, a risk model was developed, followed by the risk score (RS) calculated. Subsequently, patients were divided into high-risk group and low-risk group based on the optimal cutoff value obtained by surv_cutpoint method in survival package (version: 3.5–8) of R. In addition, KM curve was utilized to assess survival outcomes in two risk groups. The predictive performance of the prognostic model was evaluated by generating a ROC curve. In addition, a further validation analysis on model was also performed based on GSE73403. Finally, the GSEA enrichment scores for each pathway in both groups were calculated and ranked using the Benjamini–Hochberg (BH) method with P < 0.05, utilizing the clusterProfiler package in R. The top five pathways for each enrichment result were identified based on the normalized enrichment score (NES).
Immune infiltration analysis
The CIBERSORT methodology [25] was applied to investigate the infiltration levels of 22 types of immune cells within the tumor samples. To unveil differences in immune cell compositions in different risk group, the ESTIMATE analysis was employed [26]. Moreover, the link between key genes obtained above and different immune cells, as well as among different cell types, was assessed using the Pearson correlation coefficient [27]. The results were visualized using heat map. Furthermore, the expression levels of key immune checkpoint genes, such as CD274, CTLA4, HAVCR2, LAG3, PDCD1, and LMTK3, were obtained, and the differences in expression between different risk groups were evaluated through an t test.
Drug sensitivity analysis
The oncoPredict package in R was applied to explore the sensitivity of various drugs [28]. The difference of sensitivity (IC50 value) for chemotherapy drugs was quantified by using the calcPhenotype algorithm in TCGA-LUSC dataset. Significant differences in drug sensitivity between different risk groups were assessed using the t test. The result was visualized using box plot.
Cell culture
BEAS-2B (SUNNCELL, SNL-203), a human lung epithelial cell line, as well as three LUSC cell line including NCI-H2170 (Pricella, CL-0394), NCI-H520 (Pricella, CL-0402) and NCI-H226 (Pricella, CL-0396), was kept in RPMI-1640 medium (Pricella, PM150110), which further mixed with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin until they reached 90% confluence (at 37 °C and 5% CO2). All cells were cultured consistently between the second and third generations, and culture supernatant and cell samples were harvested for analysis after 24 h.
Lentiviral vectors construction and transfection
The sh-TLR3 (SS: GAGTTGTCATCGAATCAAA; AS: TTTGATTCGATGACAACTC), which designed based on Designer of Small Interfering RNA website, was introduced into lentiviral plasmid vectors. In addition, oe-TLR3 was designed by NCBI website. In summary, lentiviral shuttle plasmids and their associated packaging plasmids were generated, and high-purity, endotoxin-free plasmids were isolated using a reagent kit (Tiangen Biochemical Technology Co., Ltd., Beijing). These plasmids were then co-transfected into cells, while an empty lentiviral vector served as the negative control. After 18 h of transfection, the medium was replaced with complete culture medium. Following an additional 24-h incubation, the supernatant rich in lentiviral particles was collected and concentrated through ultracentrifugation. The viral titer was assessed using a fluorescence counting method, and the packaged virus was utilized for subsequent experiments. For lentiviral infections, NCI-H226 cells were kept in a 6-well plate, and lentivirus (1 × 10^8 TU/mL) was added to the medium when the cells reached 70–90% confluence. Following 48 h for infections, 2 µg/mL puromycin was added to stable transfected cells.
qRT-PCR
To explore the mRNA expression of signature genes (CHEK2, PDK4, TLR3 and IL1β) identified in this study, the qRT-PCR assay was conducted based on each cell lines. Total RNA was extracted from the cells with TRIZOL reagent (Invitrogen, 15,596,018), and cDNA was synthesized using the 5 × FastKing-RT SuperMix (TsingKe, KR118-02). PCR analysis was achieved by a RT-qPCR instrument (Bio-Rad, CFX96Touch) with an initial denaturation step at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 12 s and annealing/extension at 60 °C for 40 s. Relative expression levels were determined using the 2−ΔΔCt method [29]. Primer details are provided in Supplementary Table 1.
Western blotting
The western blotting assay in current study was used to evaluate the expression of CHEK2, PDK4, TLR3 and IL1β. Briefly, total protein was obtained based on cells in each group with the help of RIPA lysate (Beyotime, P0013B). Bicinchoninic acid (BCA) protein quantitative kit Solarbio (PC0020) was used in current study for the quantitative investigation. Following denaturation, the proteins were isolated with the help of 10% SDS-PAGE electrophoresis (Beyotime, ST628) and subsequently moved to a polyvinylidene fluoride (PVDF) membrane (Beyotime, FFP24). At ambient temperature, the cellular membrane was sealed with a TBS solution containing 5% BSA for 1 h. before adding primary antibody including Anti-CHEK2 antibody (Abclonal rabbit monoclonal, 1:2000, A19543), Anti-p-IL1β antibody (Abclonal rabbit monoclonal, 1:1000, A22257), Anti-TLR3 antibody (Abcam Rabbit polyclonal, 2 g/mL, ab62566), Anti-PDK4 antibody (Abclonal Rabbit polyclonal, 1:1000, A13337) and Anti-GAPDH antibody (Affinity Rabbit polyclonal, 1:3000, AF7021) used at manufacturer-recommended dilutions. Subsequently, sections were mixed with the Affinity goat anti-rabbit IgG H&L (HRP) secondary antibody (1:2000, ab205718) for 60 min at room temperature. Protein bands were then developed using ECL reagents, followed by images capturing.
MTT assay
The proliferative ability of cells was investigated by MTT. Simply, according to the instruction of MTT cell proliferation and cytotoxicity detection kit (Beyotime, C0009S), a total of 10 μl MTT (5 mg/mL) was seeded in each well. After 4 h of culturing, the medium was removed, and 100 μl Formazan solution was added into each well. Optical density (OD) at 570 nm was detected by a detector (Biotek, Synergy HTX).
Wounding healing assay to detect cell migration
Wounding healing assay was carried out to elucidate the cell migration. All cells were seeded onto 6-well dishes (5 × 105 cell/well) and wounded using a marker pen. Detached cells were eliminated by PBS washing on samples, followed by incubated in an incubator (37 °C, 5% CO2). The plates were photographed at 0 h, 24 h and 48 h, respectively.
Colony formation assay
Following lentivirus treatment, stably transfected cells were trypsinized, counted, and seeded into a 6-well plate at a density of 200 cells per well. Cells allowed to grow for 7 days to form colonies. Afterward, the colonies were stained with crystal violet (Beyotime, C0121) for 20 min. The stained colonies were imaged with a fluorescence microscope, and the overall number of colonies (50 cells each) was quantified and examined.
Enzyme-linked immunosorbent assay (ELISA)
ELISA kits were applied to explore the expression levels of immune factors including IL1β (Beyotime, PI305), TNF-α (Mlbio, ml064303), IFN-γ (Mlbio, ml057856) and VEGF (Mlbio, ml064255) in supernatant.
Statistical analysis
R software (version 4.3.3) in this study was used as tool for statistical analysis. The comparison of differences between the two groups in current study was carried out with the help of Wilcoxon rank test. In addition, the relationship between groups was carried out in our study with the help of Spearman correlation analysis. A two-sided P value of < 0.05 was considered statistically significant.






Comments (0)