
Remember me

The primary objective of the study is the percentage of participants achieving a composite of more than 50% time in range and less than 1% time below range, during the 12-week follow-up period.
Study DesignThe study will take place within primary healthcare practices in Leicester, Leicestershire and Rutland (LLR), UK. Leicester City has the highest prevalence of diabetes in England (8.9%), and a large multi-ethnic population (59.2% people of colour) [28], which is important as some ethnic minorities have up to a fivefold increased risk for type 2 diabetes compared with white Europeans [29].
This is a 12-week feasibility study to assess the impact and acceptability of a scripted medication de-intensification algorithm and HCP education alongside CGM on diabetes management. For the purposes of this study, HCPs are defined as clinical staff with prescribing responsibilities for patients with diabetes, e.g. general practitioners (GPs), practice pharmacists or nurses.
All participants will receive the study intervention and the schedule of events as shown in Table 1. This protocol has been designed using the INCLUDE Impaired Capacity to Consent framework [30] and was guided by the Mental Capacity Act [31].
Table 1 Schedule of eventsA minimum of two HCPs per practice will be required to complete the study training, which includes pre-recorded training and booster animations on the medication de-intensification algorithm (around 60 min), a 60-min pre-recorded webinar on the Dexcom ONE+ CGM and data interpretation via the Clarity platform. HCP training will be supplemented with monitoring telephone calls during weeks 2, 4 and 8 and the study team will be available with advice at any point. Additionally, HCPs will complete follow-up evaluations to assess their confidence and competency in using the algorithm and CGM. The algorithm was developed for an ongoing study (D-MED, IRAS:280971) and will guide the HCP through the decision-making process for de-intensification [32].
Additionally, a minimum of two care home staff members will receive a 60-min pre-recorded training session which covers the Dexcom ONE+ system, high and low glucose alerts and how to respond to them, how to safely dispose of the technology and how to check for skin integrity issues. Additionally, training will cover the set-up and use of the cloud-based system, Dexcom Clarity, which gathers the glucose sensing data.
Sample SelectionWe will recruit 49 participants with type 2 diabetes, aged 65 years and over living within care homes. The full inclusion, exclusion and withdrawal criteria are listed in Table 2.
Table 2 Study inclusion and exclusion criteriaMethods to recruit practices and care homes will include advertisement via the National Institute of Health Research (NIHR), direct contact, word of mouth and presentations at relevant healthcare meetings. Practices may only participate if a care home where their patients are resident also participates. NIHR ENRICH (Enabling Research In Care Homes) will distribute study information and expression of interest forms (EOI) to research-active care homes.
A database search of practice lists will be completed, and an invitation to participate, participant information sheets (PIS) and EOI forms will be mailed to eligible patients. In the event of recruitment delays, strategies such as additional outreach and extended timelines are planned.
We will recruit care home residents with and without capacity. The capacity assessment and the consent process will be guided by the Mental Capacity Act, 2005 [31] and completed by an appropriately experienced and trained research nurse or doctor.
Written informed consent will be received from participants with capacity, whilst advice will be sought, in the form of a declaration form, from consultees for participants lacking capacity. A short PIS with adaptations to language and font size is available to all participants and consultees.
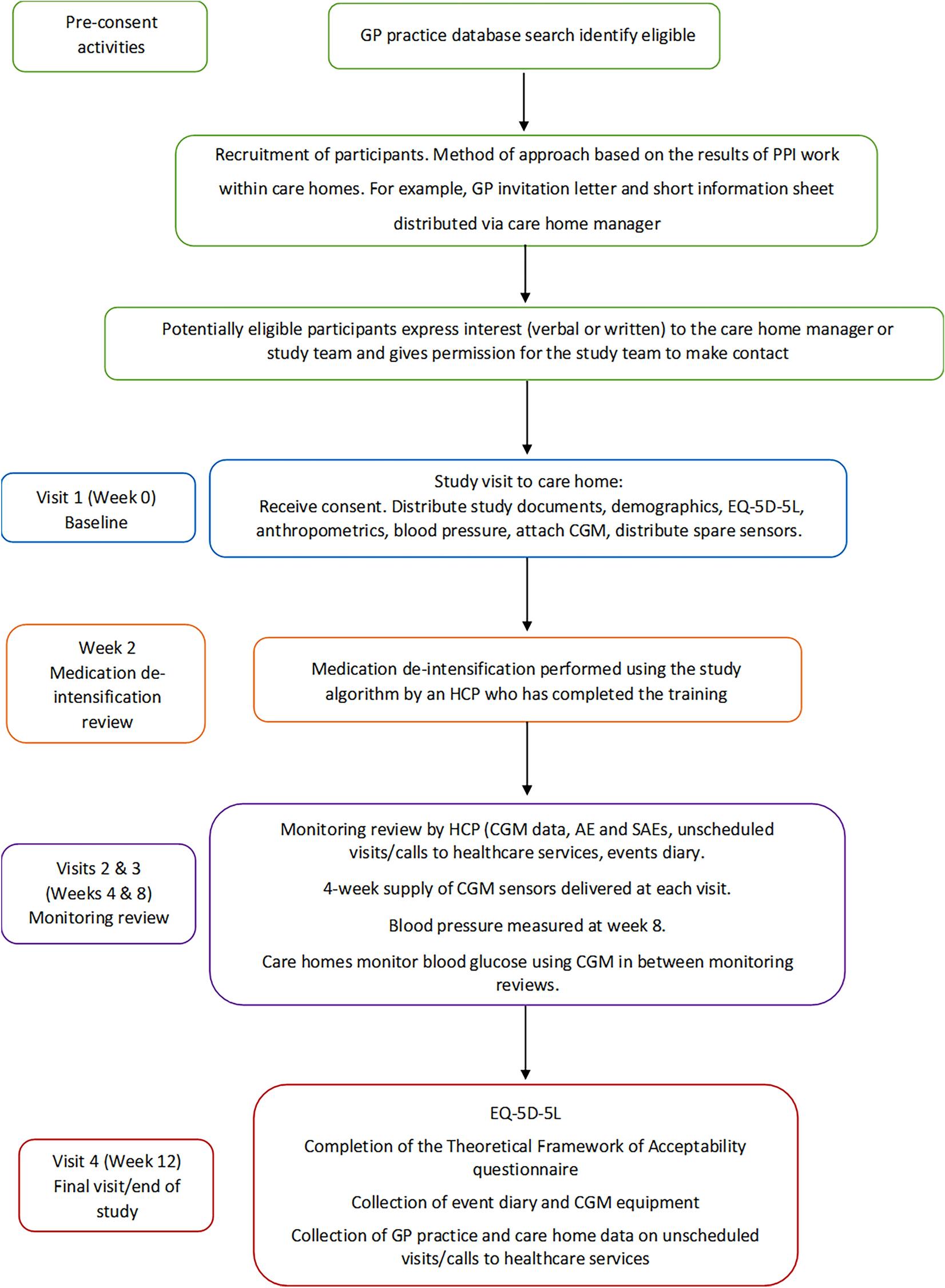
MeasurementsParticipant flow through the study is shown in Fig. 1. Participants will have four in-person study visits during the 12-week study period (weeks 0, 4, 8 and 12). HCPs will complete remote de-intensification or monitoring aided by the de-intensification algorithm, the CGM data, self-reported hypo- and hyperglycaemic events (reported by participants or their carers and shared with the practices) and any unscheduled visits to healthcare services at three points (weeks 2, 4 and 8) throughout the study.
Fig. 1
Study flow chart. AE adverse event, CGM continuous glucose monitoring, GP general practitioner, HCP healthcare professional, SAE serious adverse event
Briefly, data to be collected during the baseline visit will include a medical history, basic demographics (including age, sex and ethnicity), concomitant medications, anthropometrics including height and weight (body mass index (BMI) will be calculated), blood pressure and quality of life (EQ-5D-5L). These data will be obtained by a research nurse or member of the study team and will be repeated at the end of the study (week 12). An additional measurement of participant’s blood pressure will take place during week 8 and an additional questionnaire (Theoretical Framework of Acceptability (TFA)) will be completed with participants, their consultees and/or care home staff during week 12.
Participants will be asked to wear a real-time CGM device (Dexcom ONE+) continuously throughout the 12-week study to aid their HCP in the safe de-intensification of their medications. The Dexcom ONE+ system includes (1) an integrated sensor-transmitter that continuously measures interstitial glucose levels, (2) an over-patch for secure placement, and (3) a reader that provides real-time glucose data. Readers were chosen over smartphones for their ease of use and to prevent barriers to participation due to a lack of digital skills.
Dexcom ONE+ sensors send real-time data from the interstitial fluid to a reader every 5 minutes which can be viewed remotely by the participant’s HCP or care home staff with caring responsibilities. The sensor of the Dexcom ONE+ system needs to be changed every 10 days and will be worn on the back of participants’ arms or abdomen, as per manufacturer guidelines, and to minimize accidental removal.
The monitor can be worn during everyday activities including bathing and most physical activities. Communication between the transmitter and the reader works via Bluetooth, providing they are within a 6-m distance. Therefore, the reader will be kept with participants, ideally on their person.
Alarms signifying that the user’s glucose levels are too low/high will be set on the basis of Diabetes Technology UK guidance [33]. We will ask the care home to upload data with every sensor change to ensure practice staff have access to the data they require for the de-intensification/monitoring reviews. All CGM data will be stored securely on encrypted servers, and access will be limited to authorised personnel only.
Planned OutcomesPrimary and secondary study objectives are presented in Table 3.
Table 3 Primary and secondary study objectivesData CollectionThe primary and majority of the secondary outcomes will be measured using the Dexcom ONE+, for which data will be collected continuously throughout the 12-week study. Time above or below range will be analysed using recent recommendations on hypo- and hyperglycaemia: levels 1–3 for hypoglycaemia and levels 1–2 for hyperglycaemia [34].
Other secondary outcomes include quality of life, which will be measured at baseline (week 0) and at the end of the study (week 12) using the EQ-5D-5L [35], a valid measure for older people living in residential care [36]. Acceptability of the intervention to participants/consultees and care home staff will be assessed using the TFA questionnaire [37] at the end of the study (week 12). The TFA assesses seven domains of acceptability including affective attitude, burden, ethicality, intervention coherence, opportunity costs, perceived effectiveness, and self-efficacy. Answers are given on a scale of 1–5, with optional free text entries.
Care home staff will record hypo- and hyperglycaemic events throughout the study in a log book to include date, time and details of the event, symptoms and their estimated duration, recovery time and any action taken. Baseline demographic and medical history data will be extracted from patient medical records within the year preceding the baseline visit by a member of the practice staff at 12 weeks. Adverse events will be reported throughout the study and be responded to by members of the clinical team (e.g. GPs, diabetes specialists, or the study’s designated clinicians).
Data AnalysisOn the basis of a previous study [38], we found 37.5% of the recruited population met the primary outcome. However, the objective measures of the CGM readings, coupled with the alarms that indicate readings are out of range, means that the remaining 62.5% who are not in range at baseline will have medications reviewed to improve the time in range. It is plausible that 100% of participants will have the review of their medications with the algorithm if their blood glucose readings are out of range, but as a result of other complexities and availability of eligible participants for recruitment, 100% is not feasible.
Therefore, a more conservative effect size estimate of increment of the achievement of the primary outcome from 37.5% to 70% was chosen. Assuming 37.5% of the recruited sample will meet the primary outcome at baseline and an increase to 70% at follow-up, we will need to recruit 49 participants to show a statistically significant effect. The sample size was calculated with 90% power at a 5% significance level. The sample size was calculated in Stata using the clustersampsi command.
Participant disposition will be presented with respect to completion status, reason for non-completion, protocol deviations, and length of stay in the study. Demographic and baseline characteristics will be summarised by group. Continuous variables will be summarised as mean and standard deviation or median and interquartile range, and categorical variables will be given as counts and percentages.
For the primary outcome, a McNemar test will be used to assess the difference at baseline and week 12 in categorical variables; a paired t test will be used otherwise. Potential confounding factors will be investigated and adjusted for. All analyses will be carried out in Stata (version 15.0). No subgroup analyses or interim analyses are planned. Missing data will be managed using multiple imputation techniques. Sensitivity analyses will test the impact of missing CGM data on the primary and secondary outcomes.
Comments (0)