
Remember me
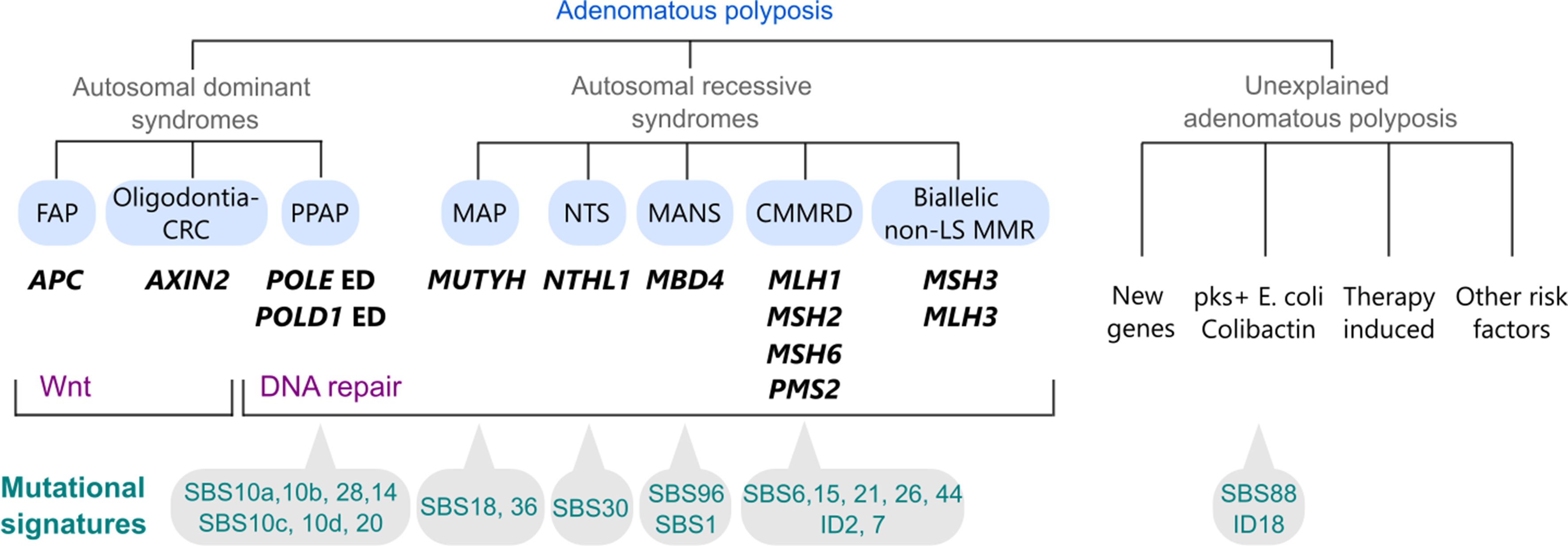




Desmoid tumors have an average annual incidence of 3.2 per million individuals [9]. The disease has a higher predilection in females with a median age of diagnosis of 38 years, and a range of diagnosis between 3 to 67 years [9,10,11,12]. The majority of DTs are sporadic in nature with about 7.5% of all desmoids occurring within a hereditary syndrome such as FAP [13,14,15]. (Fig. 1) For patients with FAP, about 12% will develop desmoid disease, with some studies reporting a cumulative risk as high as 20.6% [16, 17] The risk of DT in FAP is 852 times higher compared to the general population [18]. Both sporadic and FAP related desmoids are driven by molecular dysregulation of the WNT signaling pathway [19,20,21]. While sporadic desmoids are typically associated with stabilizing pathogenic variants in the β-catenin gene (CTNNB1) [19, 20, 22]desmoids in FAP are generally caused by deactivating pathogenic variants in the adenomatous polyposis coli (APC) gene, which in turn leads to the disinhibition and accumulation of cellular β-catenin [15, 19]. (Fig. 1) In FAP patients, and following Knudson’s ‘two-hit’ hypothesis, loss of the second APC gene leads to the development of both colorectal cancer and desmoids [23].
Fig. 1
The majority of desmoids are sporadic, and around 7.5% are associated with FAP. Sporadic desmoids are caused by CTNNB1 pathogenic variants and hereditary desmoids are caused by APC pathogenic variants
Desmoid disease represents significant morbidity and mortality in FAP patients, ranking only behind colorectal cancer and duodenal cancer. These desmoids exhibit a distinct anatomical distribution, with a higher incidence in the abdominal wall and intra-abdominal regions which anatomically account for ~ 50% of all DTs [9, 11, 13, 24]. One of the additional challenges of intraabdominal desmoid disease is the morphologic variability. Although desmoids are commonly thought of as masses, desmoid disease often manifests as diffuse, sheet-like fibrosis across the mesentery or bowel. These lesions may not show up on imaging for diagnosis. Additionally, these desmoid sheets are challenging to surgically remove as they are diffuse, infiltrating, and have poorly defined borders. Extra-abdominal desmoids may also occur in FAP, but at a much lower rate when compared to sporadic desmoids [13, 18]. FAP desmoids are more commonly observed in younger patients, with a higher male proportion, and are generally larger, more numerous, and have a greater recurrence rate compared to sporadic desmoids [13, 25]. Other risk factors for their development include prior trauma, implant site, prior radiation site or pregnancy [26,27,28]. FAP associated DT are more likely to develop at sites of prior surgical intervention and at higher rates when compared to sporadically occurring desmoid tumors [17, 25]. It is no coincidence that the majority of desmoids in FAP patients occur after abdominal surgery, when presumably a “second hit” may occur due to surgical trauma [13, 24, 29, 30].
Clinical presentation of hereditary DTs is varied. Symptoms and complications are driven by anatomic location and directly related to infiltrating mass effect on the surrounding viscera and tissues. Superficial abdominal wall desmoids present as palpable and often tender masses. Intraabdominal DTs usually present as abdominal masses and up to one third are asymptomatic [31]. These intrabdominal tumors present as mesenteric plaques or as growing intrabdominal masses. The plaques may induce mesenteric and surrounding tissue puckering, leading to bowel obstruction and hydronephrosis with subsequent renal failure. The intrabdominal masses on the other hand grow and compress intraabdominal structures leading namely to bowel obstruction, perforation with subsequent fistulae formation, intraabdominal infection and hemorrhage [11, 32,33,34,35]. Extra-abdominal desmoids can occur anywhere in the body, but are particularly prevalent around the limb girdles and proximal extremities [9, 11]. Intrathoracic DTs may compress the lung parenchyma or esophagus and cause dyspnea, coughing, and dysphagia [36, 37] Intracranial or spinal DTs are rare and manifest clinically with neurological dysfunction or pain [38, 39]. Head and neck DTs specific symptoms may include Horner’s syndrome, cervical radiculopathies and brachial plexus related neurological deficits [40].
Church et al. proposed a clinical staging system for intrabdominal and transabdominal desmoids in the setting of FAP [32, 41]. (Table 1) This system is based on four parameters including symptoms, complications, growth rate, and size [32]. Stage 1 desmoids are asymptomatic, < 10 cm and do not have any growth. Stage 2 desmoids are mildly symptomatic, < 10 cm and have no growth. Stage 3 desmoids are moderately symptomatic, 10–20 cm and are slowly growing. Complications include ureter and bowel obstruction. Stage 4 desmoids are severely symptomatic, > 20 cm and are rapidly growing. Complications include sepsis, bowel perforation and hemorrhage. These stages provide a potential framework to guide treatment options.
Table 1 Tidal efficiency along a transect with increasing distance from a tidal channel platform in 1999 and 2019. Uncertainties are standard deviationsTopics of debateDue to the rarity and variety of disease, there remains controversy, debate, and variability in the treatments for desmoid disease in FAP. The below discussion addresses some of these topics.
Role of tumor biopsy for diagnosisThere is debate regarding the need for tissue diagnosis in the management of FAP desmoid disease, depending on the clinical situation. For patients who present with an initial mass suspicious for desmoid tumor without a known FAP diagnosis, biopsy is helpful to confirm diagnosis and allows for somatic tumor testing for variants in CTNNB1 or APC. The Desmoid Tumor Working Group generally recommends biopsy for sporadic DTs [4]. Biopsy is usually performed using a core needle technique with image guidance for intraabdominal masses, or under direct palpation if on a limb or abdominal wall [42, 43]. If biopsy is not feasible, then patients are recommended to undergo germline testing for APC pathogenic variant and possibly colonoscopy to evaluate for adenomas. Risks of biopsy include bleeding and infection, although this is rare for limb or abdominal wall mass biopsies. While there is some theoretical concern that biopsy could promote further growth or seeding of desmoids in a population already at risk of developing this tumor, evidence is conflicting [4, 44, 45]. Intraabdominal lesions are often more difficult to access and can be limited by bowel or blood vessels surrounding the lesion. There is a risk of bowel injury in these circumstances. For suspected desmoid tumors in the setting of known FAP, biopsy may not necessarily be needed, particularly for mesenteric masses after colorectal surgery, where these lesions are almost always desmoids. Performing a biopsy in these cases may expose the patient to unnecessary procedural trauma without yielding substantial additional information, especially when an APC mutation has already been identified. Recent French guidelines suggest that biopsy of an abdominal or mesenteric mass after colectomy or proctocolectomy is not mandatory and should only be done if there is a strong suspicion of other etiologies [46]. Given the limited added value of biopsy and the potential risks involved, we the authors do not routinely perform a biopsy for abdominal DTs in FAP patients after colectomy.
Indications and timing of treatmentThere has been a noticeable shift in the management of DTs in the last few decades [7, 47]. It has been observed that these tumors may often spontaneously regress or experience growth arrest, leading to an indolent disease course [48,49,50]. Clinical studies have demonstrated that initial observation can yield comparable health outcomes to surgical intervention in cases of DT [49]. Consequently, non-interventional “active surveillance” is now advocated as a first line approach rather than upfront therapy [1, 4, 5, 7, 51]. This is advocated by both the Desmoid Tumor Working Group and the National Comprehensive Cancer Network (NCCN) [1, 4, 5]. Active surveillance does not impact the efficacy of subsequent interventions once indicated [4]. This strategy should include a multidisciplinary evaluation at an expert center with serial assessments and imaging (MRI or CT) performed every 3–6 months after initial diagnostic imaging for 2–3 years extended to every 6–12 months thereafter [1, 4]. A shift from “active surveillance” to “active treatment” should be considered after multiple assessments, typically at least one year after initial diagnosis, with demonstrable progression of disease or worsening symptoms [4]. Active treatment may be indicated at initial diagnosis if the desmoid disease is causing symptoms. This is particularly relevant when the tumor is involving or encroaching bowel, ureter, or head and neck which can significantly impact function, or quality of life. (Fig. 2) [1, 4, 5].
Fig. 2
Algorithm of management of desmoids in FAP patients. (QOL = quality of life)
Types of treatmentUpdated guidelines from the NCCN and the Desmoid Tumor Working Group expand the range of therapeutic options available for the “active treatment” phase of DTs [1, 4, 5]. Traditional approaches include medical therapy, surgery, and radiation. Medical therapy ranges from fairly benign treatments such as non-steroidal anti-inflammatory drugs (e.g. sulindac, celecoxib) and anti-hormonal drugs (e.g. tamoxifen, toremifene, raloxifene) to more aggressive chemotherapy including methotrexate, vinorelbine, vinblastine, doxorubicin (including pegylated liposomal formulation), dacarbazine, and hydroxyurea. When evaluating for differential effect of chemotherapeutics comparing mutational status (CTNNB1 vs. APC) no differences were detected. Notably, when comparing responses to chemotherapy based on mutational status (CTNNB1 vs. APC), no significant differences in clinical outcomes were observed [52]. Tyrosine kinase inhibitors (imatinib, sorafenib, pazopanib, nilotinib) have also demonstrated improved success, especially when compared to earlier chemotherapy regimens [53, 54]. Sorafenib is currently the only category 1 tyrosine kinase inhibitor (TKI) recommended by the NCCN for the medical treatment of desmoid tumors [1]. In a phase 3 randomized clinical trial, Gounder et al. reported an 81% 2-year progression-free survival in the sorafenib group, compared to 36% in the placebo group. However, the study did not include a subgroup analysis comparing outcomes between sporadic desmoid tumors and those associated with FAP [53]. The DESMOPAZ phase 2 trial demonstrated improved 6-months progression free survival of another TKI, Pazopanib, compared to combination methotrexate and vinblastine (83.7% vs. 45%).54 More recently, new classes of drugs have been approved for desmoid disease. Nirogacestat is a γ-secretase inhibitors and now approved by the FDA for treatment of DTs. The DeFi trial demonstrated a significant improvement in progression-free survival with Nirogacestat compared to placebo, as well as a higher probability of being event-free at the 2-year interval (76% vs. 44%).54 Outcomes remained consistent across subgroup analyses, including participants with APC somatic mutations (16% in the experimental arm vs. 15% in the placebo arm), CTNNB1 somatic mutations, and a family history of FAP. However, it is important to council female patients of reproductive age about the potential risks to fertility and the possibility of early menopause [5, 55]. In addition to these established modalities, new and emerging treatments are gaining traction. These include innovative local ablative techniques and vascular therapies like cryoablation, high-intensity focused ultrasonography (HIFU), drug-eluting bead chemoembolization, and trans arterial chemoembolization.
Although most clinicians agree that observation is appropriate for asymptomatic or mildly symptomatic desmoid disease, there is no specific guidance or consensus about initial medical treatment or escalation of therapy. The NCCN and the Desmoid Tumor Working Group give broad guidelines on navigating active therapy choice for DTs [1, 4, 5]. Particularly for FAP associated DTs, both guidelines emphasize that active treatment should be individualized and discussed in a multidisciplinary forum after a period of “active surveillance” or if earlier indications arise. In general, it is rationale to begin with a drug that is effective for the disease burden and has a good safety profile and then escalate to those with more potential side effects.
In recent years, a paradigm shift has emerged in the medical management of desmoid tumors. This is driven by the increasing availability of higher-quality evidence prompting the development of a more defined therapeutic framework. The NCCN and the desmoid working group stop short of recommending a formal medical treatment algorithm, but both bodies emphasize the importance of evidence-based decision-making, with particular attention to the level of evidence and safety profiles of the available drugs [1, 5]. NSAIDs (such as sulindac) and antihormonal therapies, once considered first-line options, are now generally avoided due to limited evidence and reliance on observational data [1, 5]. Instead, nirogacestat and TKIs, particularly sorafenib, are increasingly viewed as preferred first-line therapies. Nirogacestat, in particular, is emerging as a promising first-line agent, supported by impressive clinical outcomes from the previously mentioned Phase 3 DeFi trial and a favorable safety profile [5, 55]. Chemotherapeutics are generally considered second-line therapies. However, they become essential in cases involving symptomatic, rapidly progressing or desmoids with extensive invasion of critical structures that preclude surgical intervention. In such situations, their use is warranted due to the need for a rapid therapeutic response [57].
Surgery, when able, is an effective treatment in the correct clinical setting. Surgical resection remains the first line treatment for abdominal wall desmoids that can be safely resected [1, 4, 5]. This situation includes high likelihood of an R0 resection without functional or cosmetic compromise. Conversely, if these criteria are not satisfied, then other medical options should be first line treatment. A proposed algorithm is shown in Fig. 3. For intraabdominal desmoids, medical therapy is almost always the first consideration. Surgical resection is often challenging and not able to achieve resection due to involvement of the small bowel mesenteric root. In addition, surgery itself tends to lead to future desmoid formation. Thus, surgery should be reserved for disease that is progressive or cannot be managed with systemic medical therapy. In certain situations, such as bowel obstruction, enterocutaneous fistula, or ureteric obstruction, surgery may be the only palliative option [56]. Surgical intervention for bowel obstruction may include lysis of adhesions, intestinal bypass, or fecal diversion [56]. Surgical options for fistulae may include primary repair, bowel resection but also enteric bypass or fecal diversion. Ureter obstruction may be relieved by ureter stent or even necessitate renal auto transplantation [33]. Radiation is generally not utilized for intraabdominal desmoids due to the toxicity to surroundings structures. A proposed algorithm is shown in Fig. 4.
Fig. 3
Comments (0)