Familial adenomatous polyposis (FAP) is a hereditary cancer syndrome caused by germline mutations in the tumour suppressor gene Adenomatous Polyposis Coli (APC). These mutations predispose individuals to the ongoing accumulation of somatic loss-of-function mutations in the remaining wildtype APC allele, resulting in the development of hundreds to thousands of premalignant polyps throughout the colon and rectum over time. If left untreated, the progression to colorectal cancer (CRC) is nearly inevitable, with a lifetime penetrance approaching 100% [1]. Currently, clinical management of FAP patients revolves around prophylactic colectomy or proctocolectomy in combination with life-long endoscopic surveillance to mitigate the risk of malignant transformation [2]. Despite these interventions, the burden of disease remains significant, and there is a pressing need to explore alternative strategies aimed at cancer prevention.
Although extensive efforts have been made to identify novel chemopreventive agents, no such interventions have been successfully translated into routine clinical practice. Remarkably, most prevention approaches focus predominantly on reducing the size and number of polyps once they are formed, rather than addressing the underlying cellular and molecular processes that drive the earliest stages of adenoma development. Understanding the transition from normal intestinal epithelium to premalignant adenomas is crucial for developing interventions that could prevent adenoma formation or inhibit adenoma progression.
Given the limited availability of patient-derived samples, and the need for mechanistic investigations and high-throughput drug screening, there is a critical need for developing advanced laboratory models. These include both in vitro culture systems such as patient-derived organoids (PDO), induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs), as well as genetically engineered animal models (GEMMs). PDOs, grown from the intestinal or colonic epithelium of FAP patients retain the genetic and phenotypic traits of the original tissue, including APC mutations, making them powerful tools for studying disease progression [3,4,5]. High-throughput screening using PDOs has resulted in the identification of agents that may inhibit colorectal adenoma growth [6, 7]. However, since these PDOs lack the surrounding microenvironment that crucial for the adenoma-to-adenocarcinoma transition, culture conditions should be optimized to more closely replicate FAP pathophysiology [8, 9].
The emergence of iPSCs offers a non-invasive method for generating colonic organoids by reprogramming of patient-derived cells, mostly from the blood [10,11,12]. Similarly, ESC lines, generated from embryos following pre-implantation genetic diagnosis of FAP, can be differentiated into colonic organoids enabling the study of the earliest tumourigenic events, including APC loss [10, 13, 14]. Importantly, iPSCs and ESCs can be differentiated into multiple cell types, including epithelial, stromal, immune and endothelial cells, allowing for complex co-culture systems that better recapitulate FAP biology [15]. In addition, the use of cancer-on-chip models incorporating vascularization and immune components provide new avenues for studying disease mechanism and therapeutic testing [16,17,18].
For in vivo studies, the use of GEMMs remains indispensable. Although various animal models have been described to study the role of APC in tumourigenesis, mouse models are the most commonly used. Among these, the ApcMin model has been the most widely employed to investigate FAP. This model harbours a heterozygous nonsense mutation at codon 850, referred to as Min (multiple intestinal neoplasia), which leads to spontaneous development of 30 or more intestinal polyps by 4–6 months of age [19]. Other spontaneous models carrying mutations at different positions in the Apc gene, such as Apc1638N/+ and Apc∆716, were later developed and provided a broader spectrum of disease phenotypes [20, 21], highlighting the importance of mutation position within the Apc gene. Additionally, conditional or inducible Apc knockout models, combined with tissue-specific Cre drivers, allow researchers to study the effects of Apc mutations in specific tissues and at different developmental stages [22]. These models are often used in combination with fluorescent markers that enable the tracing of normal or mutant cell populations, or even in parallel [23]. Despite their robustness, a key limitation of these models is that the majority of adenomas arise in the small intestine, whereas in FAP patients, tumours predominantly develop in the colon and rectum. Although the underlying reasons are not fully understood, this limitation can be partially addressed in conditional models using targeted Cre drivers, such as Cdx2-Cre [24] and mCai-Cre [25] which promote adenoma development specifically in the colon.
The integration of state-of-the-art multi-omics approaches with murine or human FAP models offer a promising opportunity to study the pathophysiology of FAP more detail, thereby undoubtedly providing new insights into the earliest stages of tumourigenesis. In this review, we examine how these experimental models have expanded our understanding of neoplasia in FAP and discuss emerging strategies for cancer prevention that shift the focus from managing established polyps to preventing adenoma formation at its inception.
The earliest event in colorectal tumourigenesis: cell competition within the crypt
Given the time it takes to develop CRC, it is generally assumed that tumour initiation occurs in long-lived intestinal stem cells (ISCs) or differentiated cells that have reacquired stem-like properties. To study the behaviour of these stem cells, lineage tracing studies using genetically modified mouse models have provided crucial insights into stem cell dynamics within the intestine [26]. These models are based on the labelling of specific cell populations to allow tracking of the progeny of individual cells over time [27]. Such lineage tracing studies have revealed that, under homeostatic conditions, ISCs located at the base of intestinal crypts compete for niche occupancy. This competition is characterized by stochastic loss-and-replacement events, a process reminiscent of neutral drift dynamics [28,29,30,31]. Consequently, each intestinal crypt ultimately becomes monoclonal; inhabited by the progeny of a single stem cell. The rate at which this drift to monoclonality occurs is influenced by several factors, including the number of competing ISCs and the rate of replacement [28,29,30,31]. Interestingly, analysis of normal-appearing crypts from FAP patients showed a two-fold increase in ISCs replacements, suggesting that loss of the first APC allele might convey a selective advantage [32].
Throughout the years it has become evident that oncogenic mutations in genes such as Apc, Kras, or in other cancer driver genes, can confer a competitive advantage to mutant ISCs [33, 34]. As a result, these mutant ISCs bias drift in their favour, resulting in clonal fixation of the mutation within an intestinal crypt. Crucially, multiple studies have revealed that some oncogenic mutants, among which Apc-mutants, display ‘supercompetitor’ behaviour [35,36,37,38]. Supercompetitors do not only possess a cell-autonomous advantage, but also actively disadvantage neighbouring wildtype ISCs [39]. In case of Apc-mutant ISCs, this is reflected by the secretion of a range of Wnt antagonists, among which NOTUM is the most prominent, that result in differentiation of normal ISCs which rely heavily on Wnt signalling for their maintenance [35, 36]. Consequently, the competitive landscape within individual crypts shifts in favour of the mutant cells, facilitating the development of premalignant adenomas (Fig. 1). Importantly, genetic deletion of Notum [35] or pharmacological enhancement of Wnt signalling using lithium [36] can mitigate the competitive advantage of Apc-mutant cells and decrease adenoma burden in vivo. Together these works indicate that modulating cell competition within intestinal crypts can provide a novel strategy to prevent polyp development.
Cooperative interactions and polyclonality drive adenoma formation
Although significant progress has been made in elucidating the mechanisms underlying intra-crypt competition, how intestinal adenomas subsequently arise from these monoclonal mutant crypts remains incompletely understood. A previous study demonstrated that adenoma formation requires more than the presence of isolated Apc-mutant crypts, and only fields of mutant crypts transform into premalignant adenomas [40]. Moreover, reducing the size of mutant patches inhibited adenoma development in a non-linear manner, implying that the number of mutant crypts is not merely a probabilistic factor but instead suggests that mutant crypts may act cooperatively to facilitate tumour development [40].
Recent studies have now shed new light on this hypothesis, providing compelling evidence using state-of-the-art molecular methods that clonal cooperation is indeed required for intestinal tumour formation [41,42,43,44]. To study this phenomenon, Gaynor and colleagues used two distinct Cre-drivers to label and trace homozygous Apc-mutant ISCs at either high clonal density or as isolated mutant crypts. They revealed that, unlike the high-density clones that readily developed adenomas, the isolated mutant crypts almost never transform [41]. This work highlights the necessity for a ‘critical mass’ of Wnt mutant clones to cooperatively facilitate the development of intestinal adenomas.
Further evidence of clonal cooperation has been observed in models where Apc alleles are lost consecutively, thereby more closely resembling the pathogenesis of FAP. For example, the study by Sadien and colleagues employed a multi-colour Confetti reporter to trace clonal dynamics after heterozygous Apc loss [42]. Subsequent use of mutagenic compound N-ethyl-N-nitrosourea (ENU) led to the development of multiple lesions along the intestinal tract. Analysis of these lesions revealed a large fraction of the fluorescently labelled adenomas containing more than one colour, indicating a polyclonal origin of FAP adenomas [42]. Of these multi-coloured adenomas, around 80% carried more than two Apc mutations, and even up to 9 mutations within a single adenoma could be detected. Importantly, microdissection of individually coloured clones within the same adenoma revealed that each clone carried a distinct Apc mutation, thereby excluding the possibility of subsequent Apc mutation accumulation in a single dominant clone undergoing clonal evolution [42].These findings are supported by two additional studies leveraging advanced single-cell tracing techniques [43, 44]. Lu and colleagues used substitution mutation-aided lineage-tracing (SMALT) based barcoding [43] to uncover cell phylogenies whilst Islam et al. developed Native sgRNA Capture and sequencing (NSC-seq) that allows recording the evolutionary trajectory of individual cells as well as capturing their transcriptomic profiles [44]. Both teams implemented their tracing systems in ApcMin mice [19], that carry germline mutations in one Apc allele and spontaneously lose function of the second allele, which is the model most closely resembling human FAP. Like the work of Sadien et al., both studies demonstrated that adenomas were initiated by multiple founder clones that frequently exhibited more than two Apc mutations. Together, these studies have reported up to a hundred clones within individual adenomas, indicating a high degree of clonal diversity at the earliest stages of polyp development [42,43,44]. Importantly, this polyclonality was observed in adenoma tissues from patients with both sporadic CRC and FAP [43,44,45].
Besides the observations regarding clonal composition, all studies reported differences in behaviour between transforming and non-transforming (isolated) crypts [41], as well as between distinct clones within a polyclonal adenoma [42,43,44,45]. For example, even though isolated Apc-mutant crypts expressed known markers such as Wnt antagonist Notum [35, 36], they appeared transcriptionally more like untransformed, wildtype crypts, indicating that the full ‘adenoma phenotype’ is not achieved in single mutant clones [41]. In addition, transforming crypts demonstrated an increased accessibility of thousands of enhancers compared to single mutant crypts, which could explain the observed adenoma-specific gene regulation [41]. Interestingly, organoids cultured from the low-density (non-transforming) model do display this adenoma phenotype in vitro, suggesting a potential role for the local microenvironment in repressing this behaviour in vivo [41].
Within polyclonal adenomas, distinct clones displayed diverse proliferation rates and differentiation status, and both stem cell-enriched and fetal-like clones were reported [42, 44]. In addition, investigation of the ligand-receptor interactions revealed extensive cell-cell communication between distinct clones in polyclonal lesions, mainly involved in regulating cell adhesion and ECM composition [43, 45]. Moreover, large differences in gene expression signatures were observed in polyclonal lesions, especially influencing KRAS and MYC signalling [42, 43]. These observations all point towards dynamic, reciprocal interactions between mutant clones to collectively create ‘just right’ conditions for adenoma initiation.
These studies confirm previous observations of polyclonality in FAP [46,47,48,49,50] and reveal that a significant proportion of sporadic and hereditary intestinal adenomas are polyclonal in origin, thereby emphasizing that clonal cooperation is required during the earliest stages of adenoma formation (Fig. 1) [41,42,43,44,45]. Although the variation across these studies regarding the proportion of adenomas that were classified as monoclonal or polyclonal may reflect differences in experimental methods, it is more likely that this could be attributed to the stage of analysed adenomas. This would support a hypothesis in which adenoma progression is characterized by a gradual transition from polyclonality towards monoclonality, thereby ultimately resembling the monoclonal nature often observed in established CRCs [51].
Polyclonal-to-monoclonal transition correlates with adenoma progression
Multi-omics analyses of normal tissue, benign and dysplastic adenomas, and adenocarcinomas now provide compelling evidence that indeed points towards a model in which CRC development follows a trajectory of polyclonal to monoclonal transition [43,44,45, 52]. For instance, monoclonal lesions rarely exhibit more than two APC mutations [44], in contrast to polyclonal adenomas that are more genetically diverse [42,43,44]. In addition, patients harbouring polyclonal lesions were generally younger than patients with monoclonal adenomas, and their polyps had undergone fewer cell divisions, tended to be smaller in size, and often exhibited low-grade dysplasia [43]. On the contrary, monoclonal lesions displayed a higher degree of chromosomal abnormalities such as aneuploidy and copy number alterations and frequently harboured additional mutations in other cancer driver genes [43].
One of the largest differences was detected in the prevalence of KRAS mutations, which are significantly more frequent in monoclonal adenomas and comparable to the frequency found in established CRCs [43]. This suggests that KRAS confers an evolutionary advantage that overcomes the necessity for polyclonality and results in the selection of a single dominant clone that drives adenomas progression. Experimental evidence supports this notion as loss of Kras in combination with ENU-induced mutagenesis resulted in a larger fraction of monoclonal lesions [42]. Moreover, the correlation between the number of cell-cell interactions and the degree of polyclonality further implies that monoclonal lesions are less dependent on inter-clonal signalling [43]. Interestingly, mutations in SOX9, BCL9L and CTNNB1 are more commonly observed in early lesions than in advanced CRC, suggesting that some genes that contribute to tumour initiation eventually constrain adenoma progression [43].
In addition, Esplin et al. conducted a comprehensive analysis comparing the abundance of transcripts, proteins, lipids and metabolites between normal and adenomatous tissues from FAP patients [45]. This multi-dimensional approach revealed a plethora of signalling routes that were specifically altered as cells transition from benign to malignant lesions, particularly pathways involved in regulation of cellular homeostasis, metabolism and energy production [45]. Interestingly, these pathways were not consistently altered across all omics methods, emphasizing the importance of analysing regulatory changes across multiple dimensions [45]. Furthermore, large changes in the 3D chromatin architecture have been detected across FAP tissues with variable disease stage. Zhu and colleagues describe how, in normal cells, genes are primed for either activation or repression based on whether they display high or low connectivity between promotors and enhancers (P-E), respectively [52]. Moreover, they reveal how these connections are gradually lost during cancer progression and indicate that especially the highly connected P-E regions result in dysregulated, increased gene expression [52]. These upregulated genes were associated with cell cycle and DNA maintenance pathways [52], comparable to those found in monoclonal polyps [44]. Together, these studies reveal a complex interplay between genetic alterations, chromatin structure, and transcriptional and metabolic reprogramming that collectively drive the transition from polyclonal to monoclonal adenomas.
In addition to competing and cooperating with other epithelial clones, certain oncogenic mutants can further amplify their advantage by engaging in reciprocal interactions with the local microenvironment [37]. Although largely unexplored, it is likely that APC-mutant cells participate in similar interactions, leveraging signals from surrounding cells to promote their expansion. Recent research endeavours have particularly focused on the interactions between mutant cells and the immune system, as APC-mutant epithelial cells can elicit a plethora of immune responses, and APC mutations also influence the behaviour of immune cells. Interestingly, the earliest stages of adenoma development are characterized by a pro-inflammatory immune response, accompanied by an upregulation of the arachidonic acid pathway that mediates prostaglandin production and promotes tissue inflammation. As lesions progress towards a more malignant phenotype, this pro-inflammatory environment shifts towards immunosuppression [43,




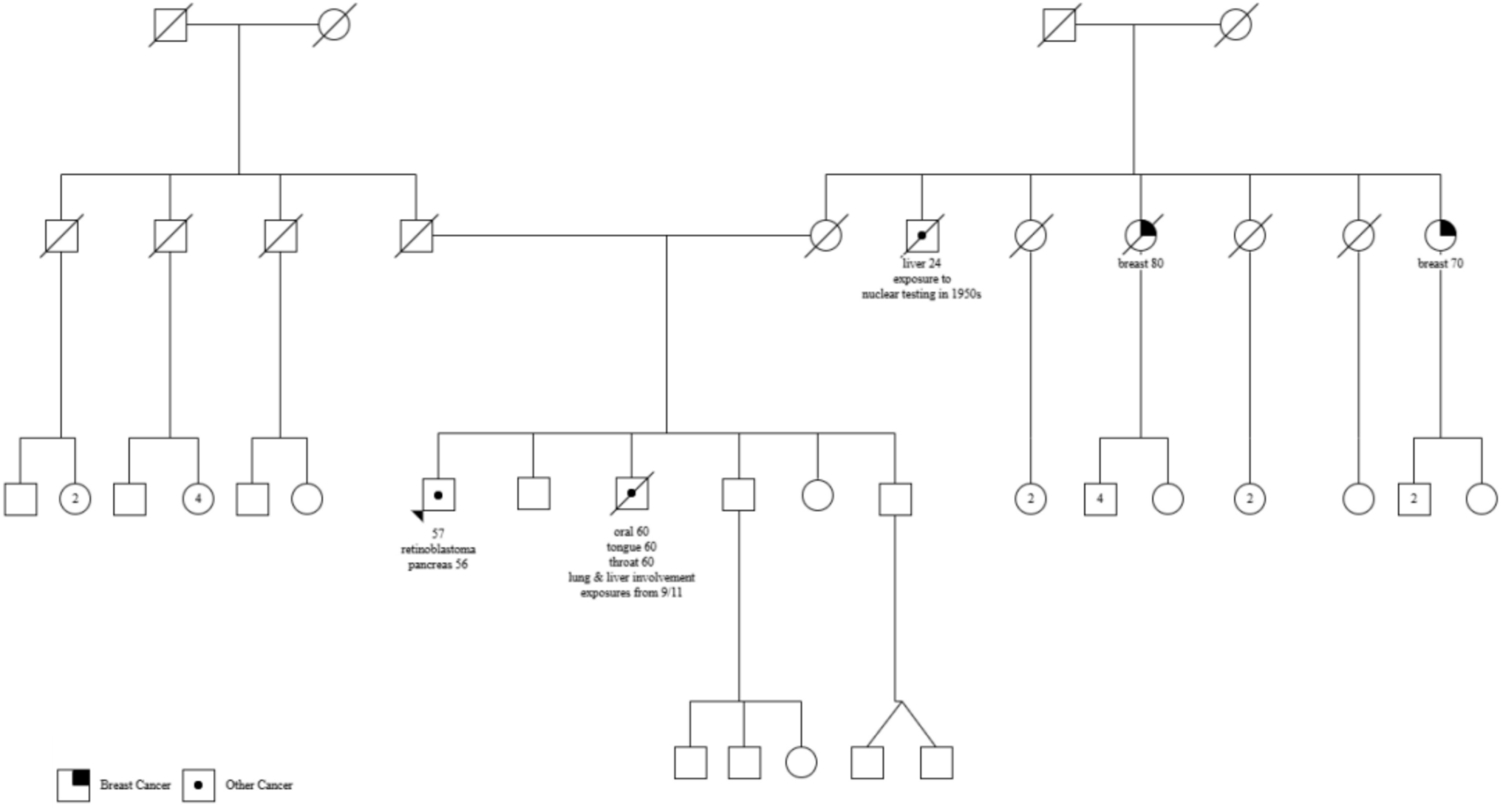

Comments (0)