
Remember me







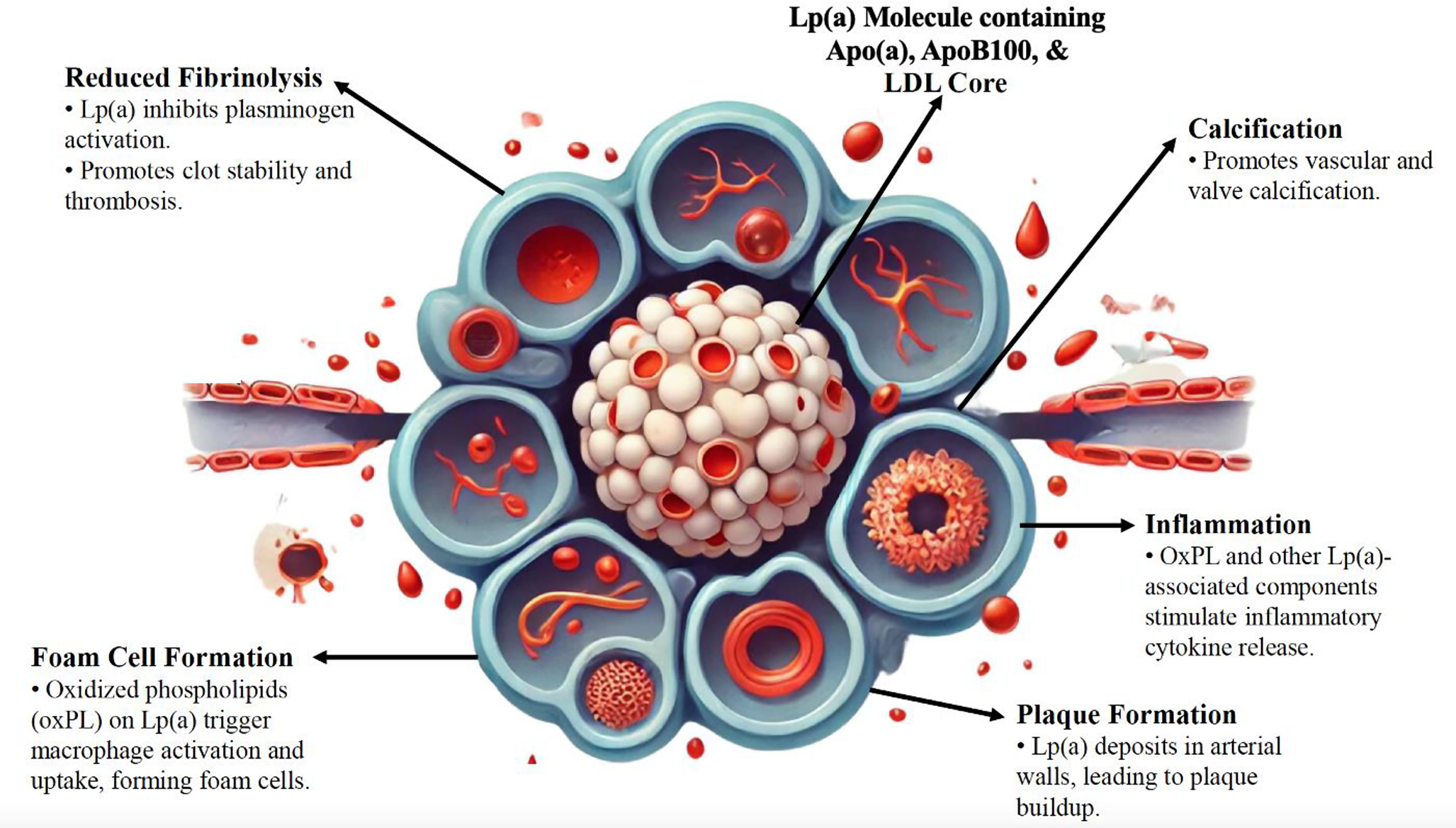
In vitro and animal studies have shown that elevated Lp(a) levels contribute to CVD risk through mechanisms such as foam cell formation, smooth muscle cell proliferation, plaque inflammation, and plaque instability, all of which drive atherosclerotic progression and plaque rupture/thrombosis [7, 8]. Notably, higher levels of Lp(a) increase CVD risk more significantly than would be expected solely from the cholesterol and apoB content contained within it, suggesting a distinct mechanism for Lp(a) in CVD [27, 28]. Potential mechanisms include the carrier role of Lp(a) for oxidized phospholipids, antifibrinolytic effects, and possible pro-platelet effects (Fig. 1) [5, 6, 29,30,31].
Fig. 1
Mechanisms Linking Elevated Lp(a) with ASCVD. Abbreviations: Apo(a), apolipoprotein(a); ApoB100, apolipoprotein B-100; ASCVD, atherosclerotic cardiovascular disease; KIV2, kringle IV subtype 2; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); OxPL, oxidized phospholipids; SNP, single-nucleotide polymorphism
Further laboratory research has identified Lp(a) as a risk factor for thrombosis. Whereas plasminogen activation to plasmin normally results in fibrinolysis of blood clots, Lp(a) does not activate plasminogen due to the inactive protease domain within apo(a) [4, 32]. In vitro and animal studies reveal that apo(a) promotes a pro-thrombotic state by inhibiting fibrinolysis through competitive inhibition of plasminogen conversion [2, 7, 8]. This has been hypothesized to provide a survival benefit by improving wound healing [3, 26]. Some studies in rabbit and human surgical samples suggest that Lp(a) accumulates preferentially at sites of tissue injury, although data on this hypothesis remain sparse [2, 3, 32,33,34]. The clinical significance of the theoretical antifibrinolytic effect of elevated Lp(a) in humans remains uncertain.
Observational StudiesSeveral observational studies have linked Lp(a) to risk for ASCVD (Table 1). The Copenhagen City Heart Study, a large prospective study that assessed the relationship between Lp(a) levels and the risk of MI and ischemic heart disease (IHD), followed 9,330 Danish participants over a 10-year follow-up period and found a stepwise increase in MI and IHD risk with elevated Lp(a) levels. Participants with Lp(a) levels > 120 mg/dL (95th percentile) had an adjusted hazard ratio (HR) for MI of 3.60 (95% CI 1.70–7.70) in women and 3.70 (95% CI 1.70-8.00) in men compared to those with Lp(a) levels < 5 mg/dL (< 22nd percentile) [35]. Additionally, levels > 120 mg/dL were associated with a 3 to 4-fold increase in relative risk (RR) of MI and absolute 10-year risks of 20% and 35% in high-risk women and men, respectively [35].
Table 1 Key Observational Studies of Lp(a) and ASCVD RiskThe ATTICA Cohort Study assessed the 10-year CVD risk in 1,890 healthy individuals from Greece with a combined endpoint of fatal or non-fatal CVD event including acute MI, unstable angina, other identified forms of ischemia, heart failure (HF), chronic arrythmias, or stroke [36]. The study found that those with Lp(a) levels ≥ 50 mg/dL had approximately twice the risk of CVD compared to those with Lp(a) < 50 mg/dL (HR 2.18, 95% CI 1.11–4.28) [<CitationRef CitationID="CR38">38</CitationRef>]. The 10-year CVD event rate was 14% in the group with Lp(a) < 50 mg/dL compared to 24% in the group with Lp(a) ≥ 50 mg/dL. The association remained statistically significant in men (HR 2.00, 95% CI 1.19–2.56) but was not statistically significant in women after adjusting for lipid markers [36].
The UK Biobank prospective study examined the relationship between Lp(a) concentrations and ASCVD risk in a larger cohort of 460,506 middle-aged participants, both with and without a history of ASCVD, over a median follow-up time of 11.2 years. The study highlighted variations in risk across different racial and ethnic groups and emphasized the significance of very high Lp(a) levels (> 150 nmol/L). A linear relationship was found between Lp(a) levels and ASCVD risk with a HR of 1.11 (95% CI 1.10–1.12) for each 50 nmol/L increase in Lp(a) [14]. High Lp(a) concentration (> 150 nmol/L) was more common in individuals with preexisting ASCVD compared to those without (20.3% vs. 12.2%, respectively). However, the RR associated with elevated Lp(a) was more pronounced in those without prior ASCVD (HR 1.50, 95% CI 1.44–1.56) compared to those with prior ASCVD (HR 1.16, 95% CI 1.05–1.27) [14].
The relationship between elevated Lp(a) and the risk of ASCVD in individuals with well-controlled plasma LDL-C levels was analyzed using the Multi-Ethnic Study of Atherosclerosis (MESA) cohort of 4,585 individuals who were all free of ASCVD at recruitment and were not on statin, fibrate, or niacin therapy [37]. The time to coronary heart disease (CHD) events, defined as MI, resuscitated cardiac arrest, and death related to CHD, was recorded over a mean follow up of 13.4 years. Participants were categorized into four groups according to the combination of LDL-C levels (≤ 100 mg/dL vs > 100 mg/dL) and Lp(a) levels (< 50 mg/dL vs ≥ 50 mg/dL). When compared to those with controlled LDL-C and Lp(a) levels, elevated Lp(a) (≥ 50 mg/dL) correlated with significantly increased CHD events, even with optimal LDL-C levels (HR 1.83, 95% CI 1.02–3.27) [37]. However, the study did not show increased risk of CHD in those with elevated LDL-C when Lp(a) was < 50 mg/dL. This study highlights how Lp(a) can contribute to ASCVD risk, even when LDL-C levels are considered optimal.
The Atherosclerosis Risk in Communities (ARIC) study sought to identify risk factors for atherosclerosis and ASCVD in diverse communities. In 3,467 Black individuals and 9,851 White individuals, an increased number of CVD events was seen at 20-year follow-up in those in the highest quintile of Lp(a) levels when compared to the lowest quintile of Lp(a) levels in both the Black population (HR 1.35, 95% CI 1.06–1.74) and White population (HR 1.27, 95% CI 1.10–1.47) [16].
Another study of 16,419 people in Boston, Massachusetts followed over a median time of almost 12 years showed an association between major adverse cardiovascular events (MACE) and Lp(a) levels [38]. In individuals without pre-existing ASCVD, those in the 71-90th and > 90th percentile of Lp(a) levels had a greater than 20% risk of developing MACE (HR 1.21 and 1.26, respectively) when compared to those in the < 50th percentile. In the group with established ASCVD, those in the > 90th percentile of Lp(a) levels had a HR of 1.93 for developing MACE compared to those in the < 50th percentile [38]. This study reiterates that regardless of whether a patient has previously had ASCVD, Lp(a) can play a role in risk for further ASCVD burden. Of course, causality is not clear in observational studies.
Recent evidence from the Women’s Health Study (WHS) strengthens the link between Lp(a) and incident ASCVD. In this large study, the association of high-sensitivity C-reactive protein, LDL-C, and Lp(a) with CVD were studied in 27,939 healthy women over a 30-year period with a composite endpoint of MI, coronary revascularization, stroke, or death from cardiovascular causes [39]. All three biomarkers independently predicted 30-year risk. For Lp(a) specifically, covariate-adjusted HR for the composite endpoint was 1.33 (95% CI 1.21–1.47) [39].
Mendelian Randomization StudiesGiven the strong genetic influence on Lp(a) levels, Mendelian randomization studies have emerged as valuable tools for assessing its causal relationship with ASCVD (Table 2). Large-scale genetic studies support this causal link. In an analysis of 40,486 participants from three Danish cohorts—the Copenhagen City Heart Study, the Copenhagen General Population Study, and the Copenhagen Ischemic Heart Disease Study—genetically determined elevated Lp(a) levels were associated with a 22% increased risk of myocardial infarction (MI) per doubling of Lp(a) levels (HR 1.22, 95% CI 1.09–1.37) [40]. Similarly, a large case-control study involving 6,497 participants (3,145 with coronary artery disease [CAD] and 3,352 controls) from the Precocious Coronary Artery Disease (PROCARDIS) Consortium identified two variants (rs10455872 and rs3798220) in the Lp(a) gene locus (LPA) linked to elevated plasma Lp(a) levels and significantly increased CAD risk (OR 1.70, 95% CI 1.49–1.95; and OR 1.92, 95% CI 1.48–2.49, respectively) [40]. These findings underscore the genetic basis of Lp(a)-mediated cardiovascular risk and provide robust evidence supporting its causal role in ASCVD.
Table 2 Genetic and Mendelian Randomization Studies on Lp(a) and ASCVD RiskLower Lp(a) levels have also been shown to have a protective effect. A Mendelian randomization study of a large, mostly Finnish population found that five splice variants at the LPA locus leading to lower Lp(a) concentrations were associated with a lower risk of CVD (OR 0.84, 95% CI 0.80–0.88) [42]. Analysis of the PROCARDIS Consortium also showed that a null allele at the LPA locus (found in about 3% of the population) was associated with a lower risk of CAD (OR 0.79, 95% CI 0.66–0.97) when compared to non-carriers [41]. Most data on CVD risk come from individuals with high Lp(a) levels so this inverse finding points to the causal effect of Lp(a) as a risk factor for CVD and provides hope that Lp(a)-lowering therapies may reduce that risk.
A Mendelian randomization analysis of the European Prospective Investigation into Cancer and Nutrition (EPIC)-CVD Consortium of 62,240 patients with CHD and 127,299 controls redemonstrated the relationship between absolute change in genetically predicted Lp(a) concentration and CHD risk [43]. Every 10 mg/dL decrease in Lp(a) concentration was associated with a 5.8% lower CHD risk (OR 0.94, 95% CI 0.93–0.95) [43]. This reduction in risk was less than the equivalent change in LDL-C, suggesting a greater magnitude of reduction would be necessary for Lp(a)-lowering therapies to be effective [43].
Meta-AnalysesSeveral meta-analyses have highlighted associations between Lp(a) concentrations and ASCVD risk, reinforcing the findings of individual studies (Table 3). In 2009, the Emerging Risk Factors Collaboration examined the relationship between Lp(a) levels and the risk of developing CHD or ischemic stroke among primary prevention populations [44]. This analysis pooled data from 126,634 participants without a history of CHD or stroke across 36 prospective cohorts. It confirmed Lp(a) as an independent, albeit modest, risk factor for incident nonfatal MI and death from CHD, with an adjusted risk ratio of 1.13 (95% CI 1.09–1.18) [44]. A higher risk was observed in participants with Lp(a) levels in the highest quartile [44]. The study also found a trend linking increased Lp(a) levels and cardiovascular events in patients who had elevated non–high-density lipoprotein cholesterol (non-HDL-C) levels [44]. While most participants were White, the study included various ethnic/racial groups, and no significant differences in risk estimates were observed across these groups.
Table 3 Meta-Analyses on Lp(a) and ASCVD OutcomesGiven the unclear clinical implications of the relationship between Lp(a) concentration and risk of ASCVD, one study sought to further clarify the impact of Lp(a) subtypes, specifically focusing on the size of apo(a) isoforms [45]. They hypothesized that smaller isoforms represent a stronger risk factor which is based on the theory that smaller isoforms are more pathogenic due to increased binding of oxidized phospholipids, higher tendency to accumulate in blood vessel walls secondary to enhanced lysine-binding and fibrin interactions, and stronger thrombogenic effect from greater inhibition of plasmin activity [50,51,52,53]. They analyzed 40 studies published between 1970 and 2009 that reported on the relationship between apo(a) isoforms and the risk of CVD and ischemic stroke, encompassing over 58,000 participants. Their analysis revealed that smaller apo(a) isoforms were associated with a two-fold increase in the risk of CHD (RR 2.08, 95% CI 1.67-2.58) and ischemic stroke (RR 2.14, 95% CI 1.85-2.97) [45]. This analysis did not determine whether isoform size was an independent risk factor, separate from Lp(a) concentration and other cardiovascular risk factors.
A 2014 paper analyzed Lp(a) levels as a prognostic biomarker for secondary prevention in individuals with CAD [46]. Plasma levels were measured in 6,708 participants with CAD from three cohorts (PEACE, CARE, and PROVE IT–TIMI 22 trials), and the data were then combined with eight previously published studies, totaling 18,978 participants. Within the three cohorts, Lp(a) levels were not associated with MACE when modeled as a continuous variable (OR 1.03 per SD, 95% CI 0.96–1.11) or by quintile (Q5:Q1 OR 1.05, 95% CI 0.83–1.34) [46]. However, when the data were combined with the other eight studies, Lp(a) levels in the highest quintile correlated with an increased risk of MACE (OR 1.40, 95% CI 1.15–1.71), although there was notable heterogeneity between studies [46]. Furthermore, a significant association was found between Lp(a) levels and MACE in those with LDL-C levels ≥ 130 mg/dl (OR 1.46, 95% CI 1.23–1.73), while no significant association was found in those with LDL-C levels < 130 mg/dl (OR 1.20, 95% CI 0.90–1.60) [46]. This meta-analysis underscored the relationship between Lp(a) levels and ASCVD in a secondary prevention population and the potential additive interplay between LDL-C and Lp(a).
Several other meta-analyses have highlighted the relationship between LDL-C and Lp(a) levels in relation to ASCVD risk. A 2018 meta-analysis of seven randomized, placebo-controlled statin trials, involving 29,069 participants, indicated a linear relationship between Lp(a) levels and cardiovascular risk, even among patients on statin treatment [47]. This meta-analysis supports the notion of Lp(a) as an independent risk factor, consistent with other recent studies showing similar results [36, 47]. Further insights were provided by a 2024 participant-level meta-analysis of six placebo-controlled statin trials, involving 27,658 participants, which emphasized the independent and additive relationship between LDL-C and Lp(a) levels [48]. In statin-treated participants, an Lp(a) level > 50 mg/dL was associated with an increased risk of ASCVD across all quartiles of LDL-C levels, including those in the lowest quartile of achieved LDL-C levels (< 77 mg/dL) (HR 1.38, 95% CI 1.06–1.79) [48]. The greatest risk was observed when both Lp(a) and LDL-C levels were in the highest quartile (HR 1.90, 95% CI 1.46–2.48), indicating an additive effect [48].
A 2024 analysis that pooled several large cohorts, including MESA, ARIC, Coronary Artery Risk Development in Young Adults (CARDIA), Jackson Heart Study (JHS), and Framingham Heart Study-Offspring (FHS-OS), encompassing 27,756 people, showed that higher Lp(a) levels are associated with an increased risk of ASCVD. Risk increased in a dose-dependent fashion with increasing risk at serum levels in the 50-75th percentile (HR 1.06, 95% CI 0.99–1.14), 75-90th percentile (HR 1.18, 95% CI 1.09–1.28) and > 90th percentile (HR 1.46, 95% CI 1.33–1.59) [49].
Comments (0)