
Remember me


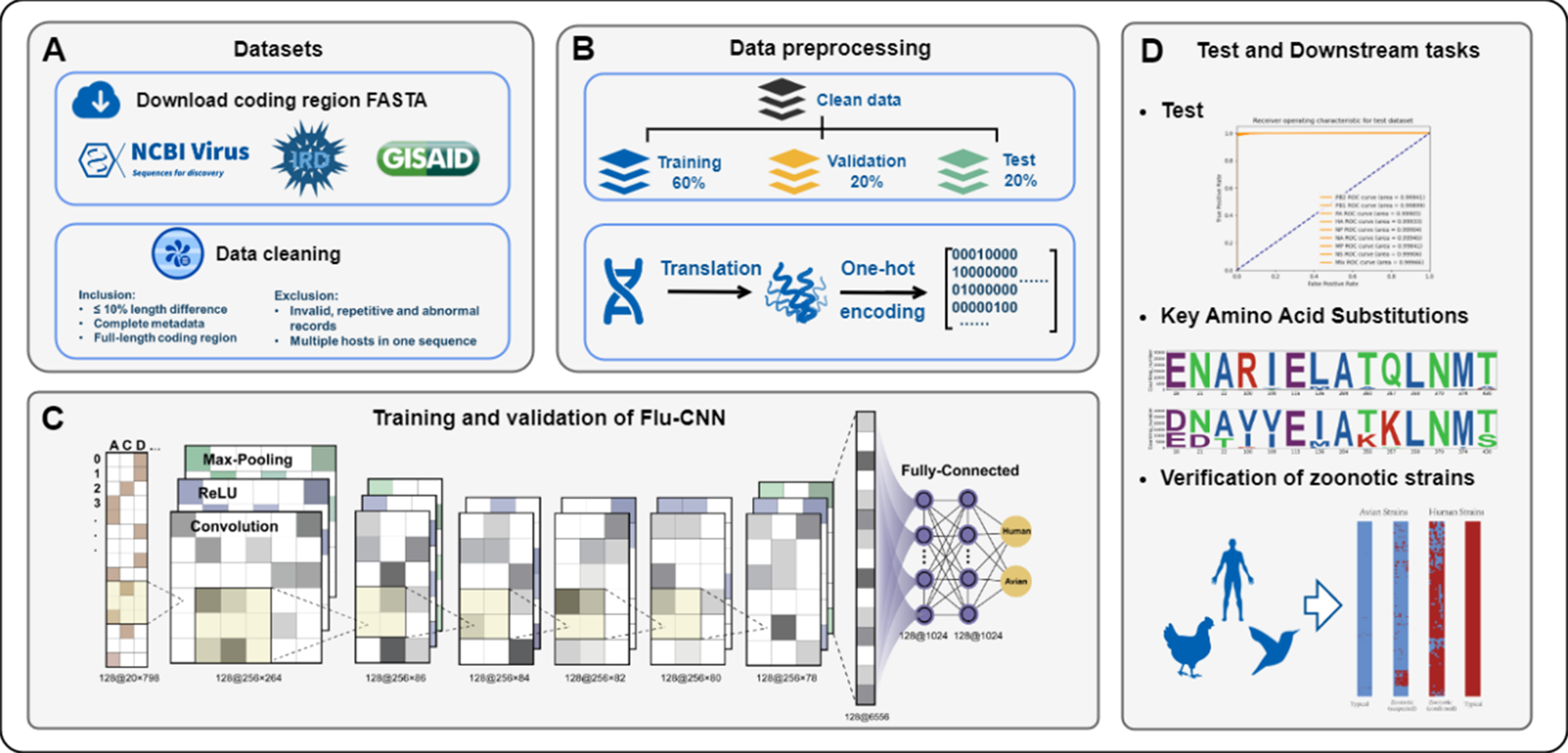
To evaluate the effectiveness of our approach in identifying IAV host specificity, we conducted a comparative analysis of Flu-CNN, ML-(d)nts, and VIDHOP. The test set included various IAV subtypes (Supplementary Table 1). FluPhenotype and phylogenetic methods were excluded due to their limitations in processing large datasets. FluPhenotype requires genome-level operations on an online platform, making it inefficient for large-scale analysis, while phylogenetic methods face challenges in constructing comprehensive trees and accurately identifying viral hosts from extensive genomic data.
The evaluation utilized four performance metrics, applied to both individual genomic segments and the entire genome (Table 1). Overall, Flu-CNN exhibited superior performance compared to other methods, both for individual segments and the whole genome. Specifically, it achieved scores exceeding 99% across all metrics for the Polymerase Basic 2 (PB2), Polymerase Acidic (PA), and Hemagglutinin (HA) segments, as well as for the complete genome. Even in the matrix protein (MP) segment, where ML-(d)nts is not applicable (as mentioned by the author [18]) and VIDHOP showed relatively lower performance, Flu-CNN maintained scores above 98% across all four metrics. Notably, Flu-CNN scored 0.9861, while VIDHOP scored 0.7600. Hence, it outperformed VIDHOP by as much as 29.8% in the MP segment.
Table 1 Performance of Flu-CNN and other methods on the test set. Mix represents segments are concatenated to assess overall performance. The ML-(d)nts method is not recommended for the MP and NS segments; thus, the corresponding results are denoted as not applicable (NA)In summary, Flu-CNN exhibits promoted accuracy in identifying IAV host specificity, outperforming established and cutting-edge methods across all four metrics, even when analyzing only a single genomic segment.
Performance across different subtypesWe further evaluated the performance of each method across different IAV subtypes. The test set was divided by subtype, and evaluations were performed on individual genomic segments as well as on genome-wide analyses across subtypes (Fig. 2).
Fig. 2
Accuracy histogram of accuracy for various methods applied to different subtypes. Each subplot corresponds to a specific subtype, with genome segments displayed on the horizontal axis and accuracy indicated on the vertical axis. Mix represents segments are concatenated to assess overall performance. The ML-(d)nts method is not recommended for the MP and NS segments; therefore, the corresponding results are marked as not applicable (NA)
In subtypes like H2N1 and H3N2, most methods performed well (Fig. 2). However, significant performance disparities emerged in subtypes such as H1N1, H2N2, and H7N9. For instance, VIDHOP may face limitations in mining and extracting information from short sequences, with its accuracy dropping below 50% in three segments (PB2, PB1, and MP) for H1N1. Likewise, in H2N2, both VIDHOP and ML-(d)nts exhibited reduced accuracy in specific segments. In contrast, Flu-CNN consistently achieved nearly 100% accuracy across all segments of H1N1 and H2N2, unaffected by these subtype-specific challenges. Particularly in H7N9, Flu-CNN showed robust and superior accuracy, outperforming other methods that struggled to reach 50% accuracy across all segments.
Despite the challenges posed by various viral subtypes that may affect the accuracy of the algorithms compared, Flu-CNN demonstrates robust and high accuracy. This consistent performance across different subtypes highlights the versatility and effectiveness of our method in identifying IAV host specificity.
Investigations on important subtypesLimited data availability for less common subtypes, such as H5N1, H7N9, and H9N2, may adversely affect performance. Despite the smaller dataset sizes for these subtypes, they pose significant risks for human infections with avian influenza. We focused on these key subtypes to evaluate the accuracy of our methods (Fig. 3). For each subtype, we randomly selected 100 sequences (50 human strains and 50 avian strains) as the test set, using the Python random module, and repeated this selection 20 times. Given the constraints of the dataset, we included four established and cutting-edge methods: ML-(d)nts, VIDHOP, phylogenetic method, and FluPhenotype. The introduction of FluPhenotype and phylogenetic methods in this section arises from their limitation in handling large-scale data. However, by sampling and extracting subtype-specific data, the dataset scale was reduced, thereby enabling the inclusion of these methods. In the context of phylogenetic analysis, predictions primarily depended on the phylogenetic tree (Supplementary Fig. 4).
Fig. 3
Ring bar chart illustrating the accuracy for various specific subtypes. Each sector area represents a genomic segment, with each point indicating an accuracy result from different methods, color-coded accordingly. The ML-(d)nts method is not recommended for the MP and NS segments and is therefore marked as not applicable (NA). The subtypes represented include: (A) H5N1, (B) H7N9, (C) H9N2
The accuracy performance of these methods for each segment of the significant subtypes is summarized (Fig. 3). VIDHOP and ML-(d)nts faced challenges in species identification for these three subtypes, achieving approximately 50% accuracy across nearly all segments. In contrast, the phylogenetic and FluPhenotype methods outperformed VIDHOP and ML-(d)nts, although they occasionally exhibited variability in their accuracy. Among the five methods evaluated, Flu-CNN consistently demonstrated the highest performance, achieving the most stable and accurate results across all segments of the three subtypes.
In summary, our evaluation of host specificity identification reveals that Flu-CNN achieves the highest accuracy overall and excels in performance across various individual subtypes. This includes critical high-risk subtypes such as H5N1, H7N9, and H9N2, despite the limited availability of sequence data for these variants. These results underscore the effectiveness of Flu-CNN in identifying the host specificity of IAV, particularly for less common but significant high-risk subtypes.
Model interpretabilityWe examined the feature representation within Flu-CNN by visualizing its intermediate convolutional layers to determine whether the model effectively captures valuable features. Specifically, we used the HA and NA segments as examples and applied Uniform Manifold Approximation and Projection (UMAP) for dimensionality reduction [27]. This technique facilitated the projection of the intermediate layer vectors into a two-dimensional space, enhancing visualization (Fig. 4).
Fig. 4
Uniform Manifold Approximation and Projection (UMAP) visualization of the convolutional layer output from Flu-CNN on HA and NA segments. Hosts and subtypes are represented by differently colored points. (A) HA segment (colored by hosts). (B) HA segment (colored by subtypes). (C) NA segment (colored by hosts). (D) NA segment (colored by subtypes)
In terms of host specificity (Figs. 4A and C), sequences exhibiting the same host specificity were clustered together, while those from different host specificity were distinguishable. This clustering indicates that Flu-CNN effectively learns essential genomic features that differentiate hosts through its convolutional layers. Regarding IAV subtypes (Figs. 4B and D), the visualized nodes were also clustered according to their respective subtypes, demonstrating Flu-CNN’s capability to extract significant informative features that distinguish between subtypes. Thus, the convolutional network of Flu-CNN captures informative features not only related to host specificity but also across various IAV subtypes.
Key amino acid substitutions for host specificityThe antigenicity of influenza virus proteins significantly influences host specificity [17]. Previous studies have identified various amino acid phenotypes that serve as candidate biomarkers for human-adapted IAVs, which are essential for understanding cross-host transmission from avian sources [26]. Therefore, the identification of human-adaptive amino acid phenotypes in influenza viruses is critical for effective surveillance and early warning systems regarding influenza outbreaks. Flu-CNN provides a computational method for the accurate identification of IAV host specificity by detecting relevant substitutions. Specifically, we screened genomic segments for amino acid mutations and employed Flu-CNN to evaluate the impact of these mutations on host specificity by assessing the resulting changes in host specificity.
By concentrating on the PB2, PA, and NP segments, we have identified key amino acid substitutions that may influence the human tropism of avian influenza viruses (Fig. 5; Supplementary Figs. 5 and 6). For example, within the PB2 protein (Fig. 5), Flu-CNN identified eight substitutions (T108V, A274S, S286G, Q591R, Q591K, E627K, D701N, D701E) that are potentially critical for host adaptability. These substitutions were on the protein’s outer surface, revealing its functional importance in PB2 structures (Fig. 5) [28, 29]. Notably, five mutations—S286G, Q591R, Q591K, E627K, and D701N—have been biologically validated as significant phenotypes influencing the human tropism of IAV [30,31,32]. The remaining substitutions (T108V, A274S, D701E) are situated in functionally significant regions: T108V resides at the N-terminal of the PB2 protein, within the minimal recognition sequence for PB1 and NP protein binding in the polymerase heterotrimer; A274S is also located at the N-terminal, associated with cap binding; and D701E is positioned at the C-terminal, alongside the D701N substitution. Although these mutations are likely contributors to host specificity, their precise effects warrant further investigation in future studies. Additional findings for the PA and NP segments are also detailed (Supplementary Figs. 5 and 6).
Fig. 5
Key human-adapted amino acid substitutions of the PB2 protein (PDB: 6QPF) screened by Flu-CNN, visualized using Visual Molecular Dynamics (VMD). Yellow indicates experimentally verified substitutions, while blue denotes substitutions not reported in the current literature. Other regions of the protein are shown in grey
This analysis demonstrates the utility of Flu-CNN in identifying critical amino acid substitutions that influence the host specificity of influenza viruses, offering insights essential for influenza surveillance and management strategies.
Identification of zoonotic IAV strainsZoonotic IAVs pose a significant global epidemic threat, which can be assessed using Flu-CNN. We utilized a manually curated dataset of zoonotic strains categorized into four groups: 5,685 typical avian influenza strains, 5,110 typical human influenza strains, 126 confirmed zoonotic strains isolated from humans, and 346 suspected zoonotic strains isolated from avian sources [15]. These groups served as labeled sequences to investigate host specificity as identified by Flu-CNN.
Predictions of host specificity for categorized zoonotic IAVs were visualized (Fig. 6). Generally, most IAVs exhibited single-host specificity (Fig. 6). Typical avian strains (Fig. 6A) and human strains (Fig. 6D) demonstrated consistent host specificity patterns, indicating nearly exclusive adaptations to avian and human hosts, respectively. In contrast, suspected zoonotic strains originating from avian sources (Fig. 6B) and confirmed zoonotic strains from humans (Fig. 6C) displayed a mosaic pattern of adaptations, encompassing both human and avian characteristics across genomic segments. The analysis of categorized zoonotic and typical IAVs revealed consistency between the results of Flu-CNN and previous studies [15]. Both Flu-CNN and previous studies [15] have demonstrated the existence of a species barrier that limits cross-host transmission of IAVs, particularly in avian strains predominantly composed of avian genes. The hyperplane representing this species barrier can be learned through the deep learning mechanisms used by Flu-CNN, highlighting the method’s effectiveness and potential applications. Furthermore, among confirmed zoonotic strains, a significantly higher proportion exhibited human tropism compared to suspected zoonotic strains. This phenomenon indicates varying zoonotic risks among strains, with confirmed zoonotic strains posing a greater risk to humans than suspected ones.
Fig. 6
Segmental host specificity signatures identified by Flu-CNN for human, avian, and zoonotic influenza strains in the previous dataset, with each row representing a strain and each column representing a genomic segment. Red indicates human adaptation, while blue indicates avian adaptation. (A) Typical avian strain. (B) Suspected zoonotic strains isolated from avian sources during zoonotic outbreaks. (C) Confirmed zoonotic strains isolated from human sources during zoonotic outbreaks. (D) Typical human strain
Fig. 7
Segmental host specificity signatures identified by Flu-CNN for human, avian, and zoonotic strains in the dataset curated for this research, with each column representing a strain and each row representing a genomic segment. Red indicates human adaptation, while blue indicates avian adaptation. A Avian strains. B Human strains
In addition to the previously categorized data (Fig. 6) [15], we utilized Flu-CNN to identify zoonotic strains within the entire dataset collected for this research (Fig. 7). While most strains adapted to a single host, seven lineages exhibited a mosaic pattern of host adaptability across four subtypes: H5N1, H5N6, H7N9, and H9N2. This phenomenon highlights the potential zoonotic risk associated with these IAV lineages for cross-species transmission. Consequently, Flu-CNN’s ability to detect such mosaic patterns underscores its crucial role and promising applications in identifying zoonotic risks in influenza viruses.
Comments (0)