
Remember me







To comprehensively dissect the spatial heterogeneity of AM, we collected biopsy samples from 3 patients, utilizing spatial features to depict the tumor heterogeneity at different progression stages. Given the high invasiveness and lethality of AM, we used microsatellites to identify the dynamic evolution of early AM invasive characteristics. Here, our analysis revealed distinct metastatic phenotypes across melanoma progression stages, from primary tumors to early metastatic lesions, representing distinct progression stages: primary non-metastatic tumors (with tumor cells confined to the epidermal layer) (Fig. 1A), metastatic lesions without microsatellites (exhibiting diffuse subcutaneous infiltration but lacking microsatellite formation) (Fig. 1B), and metastatic lesions with microsatellites (showing vertical dermal invasion and distinct tumor foci) (Fig. 1C). Following the AJCC 8th edition melanoma staging criteria for microsatellite definition, our integrated analysis using H&E staining and spatial transcriptomics (ST-seq) revealed that primary lesions contained atypical melanocytes restricted to the epidermal layer (Fig. S1A; S1B; S1C), while microsatellite-positive metastases demonstrated tumor cells invading through the dermis with discontinuous tumor-stroma boundaries (Fig. 1D; S1D; S1E). In contrast, microsatellite-negative metastases showed a pattern of diffuse subcutaneous spread without focal satellite formation. This systematic approach enabled us to delineate the dynamic evolution of early invasive features in AM progression through microsatellite patterning.
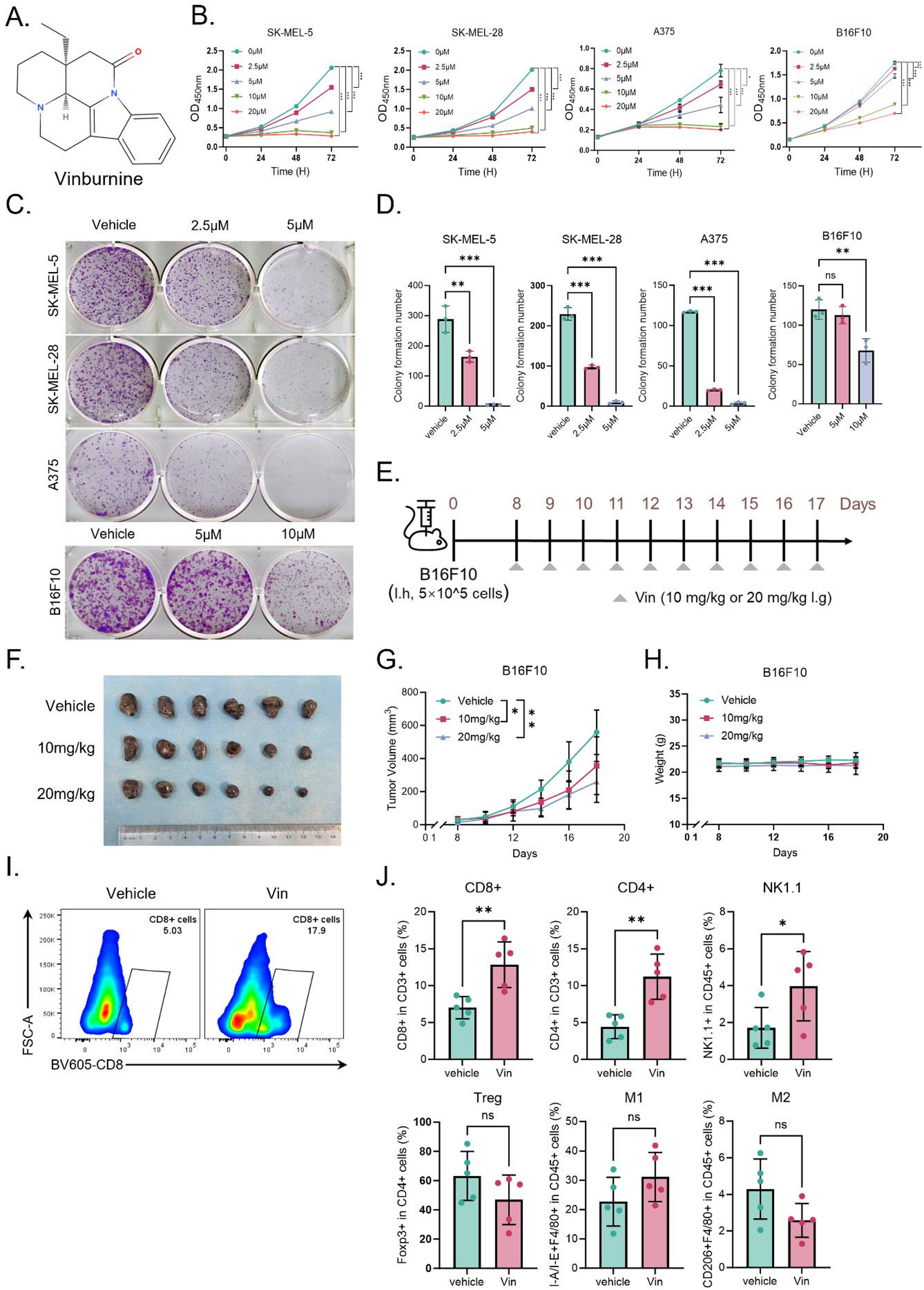
Fig. 1
Spatial transcriptomics identifies of AM. (A) H&E staining of primary AM lesion sections. The primary lesion is mainly manifested as a melanoma focus confined to the epidermal layer. Scale bars = 100 μm. (B) H&E staining of non-microsatellites metastatic lesions AM lesion sections, which exhibited diffuse subcutaneous infiltration but lacking microsatellite formation. Scale bars = 100 μm. (C) H&E staining of early microsatellite lesion sections from AM (n = 2). Early metastatic foci formed by melanoma cells vertically invade the dermis, creating a clear histological interval between the primary and microsatellite lesions. Scale bars = 100 μm. (D) Spatial transcriptomics classification of an early microsatellite AM lesion section from sample M_A. The lesion was divided into four regions through marker genes, and a clear histological interval between the primary and microsatellite lesions was identified. Scale bars = 100 μm. (E) Display of marker genes for the four categories of regions in the spatial transcriptomics of different AM lesion sections. The main epidermal and melanoma marker genes are annotated. (F) GO enrichment of marker genes for the four categories of regions in the spatial transcriptomics of different AM lesion sections. Biological processes were selected based on the count of gene number (gene count > 10) and P value (P value < 1 × 10− 2). (G-H) Upregulation of the “MYC target gene” pathway and downregulation of the “antigen presentation” pathway are displayed using spatial transcriptomics. Immuno-suppressive features was identified in the microsatellite lesion. Scale bars = 100 μm. (I) The results of stemness scoring are displayed using spatial transcriptomics. A distinct tumor adaptive phenotype was identified in the microsatellite lesion. Scale bars = 100 μm
For further insight, we applied ST-seq technology using the 10X Genomics Visium platform. After strict quality control, the two microsatellite slides, possessing a total of 6494 spots, were analyzed, with the median sequencing depth of a single spot of about 8920 unique molecular identifiers (UMIs) and 3236 genes. Next, we used unbiased clustering analysis to characterize the spatial diversity and classified the spots of the tumor sections based on the expression of epithelial, stromal, and melanoma marker genes. We manually checked the clusters according to the anatomical locations depicted by H&E staining and finally annotated four main regions: (1) primary tumor, (2) stroma, (3) microsatellite metastatic lesion, (4) epidermis (Fig. 1D; S1E). The H&E images and corresponding ST-seq spots mutually validated each other, confirming our spatial transcriptomics-based classification .
Further analysis of the marker genes of each region showed that the primary lesions were enriched for pathways related to “regulation of leukocyte differentiation”, “extracellular matrix organization”, and “immunoglobulin-mediated immune response” (Fig. 1E). In contrast, the microsatellite lesions were characterized by increased expression of pathways involved in RNA synthesis and gluconeogenesis regulation processes, even mitochondrial function, indicating enhanced transcriptional activity and energy metabolism in the microsatellites, which is consistent with stronger tumor invasiveness (Fig. 1F). Additionally, the microsatellite lesions displayed the upregulation of “MYC target” pathways and downregulation of the “antigen presentation” pathway (Fig. 1G and H; S1F; S1G). In addition, the stemness score of the microsatellite lesions was higher than that of the surrounding areas, implying a more malignant and adaptive tumor subpopulation (Fig. 1I). Collectively, our findings suggest that the presence of melanoma microsatellite lesions is associated with distant invasiveness consisting with previous studies [21].
Multifunctional melanoma cells contribute to the development of satellite lesionsWhile the initial spatial results revealed the localization and transcriptional differences between microsatellite foci and primary lesions, each spot in ST-seq contains a microscopic anatomical area with multiple cells. Therefore, to further characterize the microsatellite lesion-associated cell population, we carried out scRNA-seq analysis.
After standard processing and quality filtering of the original sequencing data, a total of 16,815 cells were retained for analysis. Unsupervised, graph-based clustering and UMAP visualization showed 17 different cell clusters, which were further annotated into 12 major cell types, including melanoma cells (C1-4), secretory cells (C5), epithelial cells (C6), epidermal cells (C7-8), VSMC (Vascular Smooth Muscle Cell) (C9), pericytes (C10), endothelial cells (C11), fibroblasts (C12), T cells (C13-14), macrophages (C15), plasma/B cells (C16), and mast cells (C17) (Fig. 2A-C).
Fig. 2
Single cell transcriptome reveled multifunctional melanoma cells in AM. (A) Single-cell annotation of two samples, with a total of 17 clusters annotated for 12 major cell types. (B) UMAP visualization showing the origin of annotated cells at different stages. (C) Cell type annotation using existing marker genes across the 17 major cell populations. (D) Stacked bar plot showing the proportion of each cell type in primary lesions and microsatellite lesions. (E) Display of marker genes and GO enrichment pathways for the top genes of the four melanoma cells. (F) Spatial locations of the four melanoma cells corresponding to gene expression profiles, demonstrating the complexity of the four cell types and the interrelationship among cells. (G) Spatial locations of immune cells corresponding to gene expression profiles, illustrating the distinct immune cell exhaustion within microsatellite lesions. (H) Violin plot displaying CNV scores across 17 major cell types, with C1 exhibiting the highest CNV burden
Compared to primary lesions (~ 10% melanoma cells), microsatellite lesions demonstrated a significantly elevated melanoma cell proportion (> 60%, clusters C1-C4), suggesting the critical roles of melanoma cells in microsatellite pathogenesis (Fig. 2D). For additional insight into the functions of the 4 main melanoma cell populations (C1-4), we performed GO analysis for top differentially expressed genes identified in these melanoma cells (Fig. 2E). Interestingly, the 4 clusters exhibited distinct characteristics indicating complex heterogeneity within and between tumors. Cluster C1 was enriched for “developmental cell growth”, “stem cell differentiation”, and “pigmentation” pathways, primarily localized to the microsatellite lesions, suggesting a role in promoting melanoma cell maturation. Cluster C2 was enriched for “developmental pigmentation”, “melanin biosynthesis”, and “secondary metabolic processes”, potentially representing the main melanin-secreting cells. Cluster C3 was enriched in “cytoplasmic translation”, “oxidative phosphorylation”, and “detoxification” processes, which evidenced by elevated S100A1/S100B and suppressed proliferation markers, and Cluster C4 was enriched in genes related to “DNA replication” and “cell cycle processes”.
To resolve cellular composition in spatial transcriptomics, we employed the RCTD (Robust Cell Type Decomposition) algorithm to deconvolve spot-level expression data using single cell-derived reference profiles. This approach enabled quantitative mapping of cell type proportions for each visium spot. The C2 cells were significantly co-localized in both primary tumors and microsatellites. In contrast, C1 and C3 cells were mainly concentrated in microsatellites (Fig. 2F; S2A). We also mapped other cell types, such as endothelial cells, epithelial cells, and keratinocytes, to their anatomical locations (Fig. 2G; S2C). Fibroblasts were more prevalent in non-microsatellite areas, interwoven with other types of cells around the microsatellites (Fig. S2B; S2C). Consistent with H&E staining patterns, these results suggest spatial co-localization of fibroblast populations and vascular components. Importantly, compared to the immune response of the primary lesions, barely any immune infiltration was detected inside the microsatellite lesions. T cells, B cells, and macrophages could only be detected outside the microsatellite lesions, consistent with the downregulation of “antigen processing and presentation” pathway identified in the spatial transcriptomics analysis. These findings illustrated the immune cell exclusion in microsatellite metastatic lesions, which are consist with highly immunosuppressive microenvironment of LN metastasis reports relevant with invasive potential and poor prognosis [5].
To further characterize the cell subtypes, we inferred the CNV levels in each cluster, according to average expression patterns across intervals of the human genome [22, 23]. The highest level of CNV, which plays an important role in the pathogenesis and poor prognosis of tumor patients, was observed in melanoma cells, especially cluster C15 (Fig. 2H; S2D). These results, together with the functional annotation results, highlight the crucial roles of the melanoma cell clusters, C1 and C2, in malignant progression.
Expression characteristics of highly invasive melanoma subpopulationsGiven that melanoma cells manifest multiple and/or overlapping/hybrid phenotypes [9] we performed subclustering analysis of the four melanoma cell clusters and examined the gene expression features (Fig. S3A). The results indicated that C1 and C2 clustered together, while C3 and C4 were separated into distinct clusters, consistent with the overarching function of C1 and C2 in melanoma maturation.
Based on the invasiveness of AM, we first utilized the melanoma-specific gene signature “ALONSO_METASTASIS_UP” from the MSigDB to parse the effects of different melanoma cells on tumor metastasis (Fig. 3A) [24]. Compared to C3, C1/C2/C4 subpopulations showed higher metastasis feature, including melanoma invasion marker genes MITF and PMEL, which were highly expressed in C1/C2 (Fig. 3B; S3B). Subsequently, we conducted activity (AUC) scoring using single-cell data. Violin plots indicated that “fatty acid metabolism” and “fatty acid transport” were high in all melanoma cells. Furthermore, the “short-chain fatty acid metabolism” pathway was specifically enriched in the C2 subpopulation, suggesting an association between AM invasiveness and fatty acid metabolic dysregulation (Fig. 3C). This dysregulation likely promotes AM invasiveness through C1/C2-mediated mechanisms rather than via C3 – an metastasis-negative cluster lacking MITF expression [5].
Fig. 3
The funtional detcetion of melanoma cells. (A) The melanoma-specific gene signature ‘ALONSO_METASTASIS_UP’ (MSigDB) was employed to assess metastatic potential across the four cell subtypes, demonstrating elevated signature scores in C1, C2, and C4. (B) Spatial localization and expression profile detection of MITF in various cell types. (C) Pathway activity scoring (AUC Score) for “fatty acid metabolism” and “fatty acid transport”, with “short-chain fatty acid metabolism process” showing specifically high levels in C2 subpopulation. (D) Identification of complex intercellular communication in AM microsatellite lesions through CellChat analysis, with a focus on the “NOTCH/NECTIN”, “GAS/TNF”, and “TGFβ/WNT” pathways. (E) Schematic of the SOX6 screening workflow. Upregulated candidate genes were intersected between ST-seq and scRNA-seq data, followed by transcription factor (TF) screening and clinical validation using TCGA survival data. (F) Spatial localization of SOX6, indicating specific high expression in microsatellite lesions. (G) Violin plot showing SOX6 expression distribution across four melanoma cell subtypes. (H) Box plot showing SOX6 expression in primary and metastatic tumors in TCGA database. (I) Detection of SOX6 protein levels in primary and lymph node (LN) metastasis samples (n = 6). Scale bars = 100 μm. (J) Detection of SOX6 protein levels in primary and microsatellite metastasis samples (n = 6). Scale bars = 100 μm
We also used CellChat to detect intercellular communications in the microsatellite lesions (Fig. 3D). As signal senders, C1/C2 and even C4 subpopulations were observed to promote tumor-related cell type switch through the Notch/NECTIN signaling pathway, which could facilitate tumor migration. C1/C2 cells were also predicted to determine the fate of cancer-associated fibroblasts through the Notch pathway, thereby regulating the tumor microenvironment. The results are consistent with the possibility that C1/C2/C4 cells, as senders of NECTIN signals, mainly transmit signals to T cells, regulating immune processes in tumors. In addition, melanoma subpopulations were detected as receivers of immune signals such as GAS and TNF. However, the CD226 signaling pathway was involved in intercellular signal communication in the C2/C4 subpopulations in primary lesions, while no CD226-NECTIN signal interaction was detected in the melanoma subpopulations in microsatellite lesions. This could potentially explain the ineffectiveness of immune checkpoint blockade therapy in the metastatic foci of melanoma (Fig. S3D). Notably, well-known melanoma pathways, such as TGF-β and WNT, were found to mainly affect the C1 melanoma cell subtype through intercellular communication. These results indicate that epidermal tumor-promoting signals drive the transformation of C1 cells, which is consistent with the higher CNV scores observed in the C1 cluster. Integrating the localization and functional analysis of cell invasiveness of the four melanoma cells, we consider C4 to be the primary lesion in the epidermal layer. While the MITF-negative C3 cluster exhibits upregulated S100A1/S100B with downregulated proliferation markers and absence of cellular interactions, displaying a “silent” phenotype as potentially reflecting immune evasion or dormancy mechanisms [5]. With higher CNV scores and intercellular communications, C1/C2 were identified as the main driver subpopulations promoting tumor invasiveness.
To elucidate the transcriptomic features and oncogenic mechanisms of driver subpopulations promoting early metastasis, we integrated differentially expressed genes (DEGs) from microsatellite spatial transcriptomics with those from the four melanoma cell clusters (Fig. 3E). Specifically, genes upregulated in spatial transcriptomic (ST) microsatellite lesions (log2FC > 0.2; p_value_adj < 0.001) and genes upregulated in scRNA-seq C1/C2 clusters versus C3/C4 (log2FC > 0.2; p_value_adj < 0.001) were intersected. This identified 93 genes enriched in pathways including “negative regulation of immune system process”, “negative regulation of cell population proliferation”, and “developmental pigmentation” (Fig. S3E). Among these, we detected co-upregulated melanoma marker gene MLANA and metabolic genes FASN, GPX3, and APOD, suggesting metabolic reprogramming as a key factor in invasiveness. Functional annotation using DisGeNET revealed significant association of these 93 genes with metastatic melanoma (p = 3.16 × 10− 5), predicting their collective role in driving AM metastasis through differential expression in C1/C2 cells (Fig. S3F) [25].
Moreover, key transcription factors (TFs) were screened to identify the AM invasiveness driver. 10 TFs were selected form the 93 candidates, such as, HIF3A, MYC. Prognostic analysis via TCGA database highlighted three TFs with significant negative survival impact (Fig. S3G; S3H). Given the established role of SRY-box (SOX) TFs (e.g., SOX4, SOX9, SOX10) in AM metastasis [26,27,28,29] we focused on SOX6 - highly expressed in ST microsatellite lesions, elevated in C1/C2 populations, and associated with poor prognosis (p = 0.00063) (Fig. 3F and G; S3C). TCGA analysis even confirmed SOX6 upregulation in cutaneous melanoma (SKCM) (Fig. 3H). Distinct high-expression of SOX6 was also observed in additional biopsy samples, implicating its functional role in both AM microsatellite and even lymph node (LN) metastasis (Fig. 3I and J).
Collectively, these results demonstrate that C1/C2 melanoma cells with elevated SOX6 expression drive tumor invasiveness.
High expression of SOX6 promotes cancer cell invasionTo verify the role of SOX6 expression in promoting tumor invasion in AM, we established SOX6-overexpressing (SOX6_OE) A375 and SK-MEL-28 stable cell line via lentivirus transduction and checked the cell proliferation and invasive capabilities. Through PCR and Western blotting assays, we confirmed that the transcription and protein levels of SOX6 in the SOX6_OE cell line were significantly up-regulated (Fig. S4A; S4B). SOX6-Knockout (SOX6_KO) A375 and SOX6-Knockdown (SOX6_KD) SK-MEL-28 stable cell lines were also used to validate the role of SOX6 in AM invasiveness. Using PCR and Western blotting assays, SOX6 were near-complete loss in SOX6_KO/SOX6_KD cells (Fig. S4C; S4D).
Subsequent functional validation under physiologically relevant hypoxic conditions (present in > 90% of solid tumors) [30] revealed that SOX6 overexpression (SOX6_OE) significantly enhanced cellular invasiveness (Fig. 4A). Following 48-hour hypoxia exposure, SOX6_OE cells exhibited elevated proliferation rates and accelerated wound closure in scratch assays, indicating potentiated migratory capacity (Fig. 4B and C). Conversely, SOX6 knockout (SOX6_KO) in A375 and SOX6 knockdown (SOX6_KD) SK-MEL-28 melanoma lines demonstrated reciprocal phenotypes: markedly impaired invasiveness, diminished migration, and reduced proliferation (Fig. 4D-F). These complementary in vitro experiments collectively establish SOX6 as a critical regulator of melanoma cell proliferation, migration, and invasion.
Fig. 4
In vitro and in vivo validation of SOX_OE promoting cancer cell invasion. (A) Evaluation of the invasive capacity of SOX6_OE A375 cells under hypoxic conditions. (B) Evaluation of the migratory capacity of SOX6_OE A375 cells under hypoxic conditions. (C) Evaluation of the proliferative capacity of SOX6-overexpressing A375 and SK-MEL-28 cells under hypoxic conditions. (D) Evaluation of the migratory capacity of SOX6_KO A375 cells. (E) Evaluation of proliferative capacity of SOX6_KO A375 and SOX6_KD SK-MEL-28 cells. (F) Evaluation of the invasive capacity of SOX6_KO A375 cells. (G) Detection of the pro-tumor invasive ability of SOX6 overexpression in A375 cells by intravenous tail injection in mice (n = 5). (H) H&E staining of lung metastasis in mice injected with SOX6_OE A375 cells via the tail vein. Scale bars = 40 μm. (I) Detection of the pro-tumor occurrence and development ability of SOX6 overexpression in A375 cells by subcutaneous injection in mice (n = 5). (J) Detection of SOX6 protein levels in lung metastases of mice injected with SOX6_OE A375 cells via the tail vein. Scale bars = 100 μm. (K) Detection of KI67 protein levels in lung metastasis of mice injected with SOX6_OE A375 cells via the tail vein. Scale bars = 100 μm
Further in vivo validation using a tail vein injection model with SOX6_OE A375 cells in nude mice confirmed that SOX6 overexpression drives tumor progression and metastatic dissemination. After four weeks, the lungs of mice injected with SOX6_OE cells exhibited a significantly number of increased metastatic lesions [44.0 ± 9.6 vs. 12.6 ± 8.0 (tumors per lung; p < 0.01); (Fig. 4G)]. Histological examinations of lung tissues confirmed increased tumor size in the SOX6_OE group (Fig. 4H). Mice subcutaneously injected with the SOX6_OE also developed significantly larger subcutaneous tumors [0.10 ± 0.02 g vs. 0.02 ± 0.03 g (tumors per subcutaneous; p < 0.01); (Fig. 4I)]. Immunohistochemical analysis further corroborated the overexpression of SOX6 and KI67 in both pulmonary and subcutaneous SOX6_OE tumors (Fig. 4J and K; S4H). These results demonstrate that SOX6 overexpression facilitates melanoma progression and enhances metastatic potential, underscoring its role as a critical regulator in melanoma.
High expression of SOX6 regulates metabolic reprogramming to promote tumor developmentTo further explore the downstream mechanisms of SOX6 in tumor metastasis, we performed transcriptome sequencing to compare gene expression profiles between i) SOX6-overexpressing (OE) vs. empty vector (Mock) A375 cells; and ii) wild-type (WT) vs. SOX6-knockout (KO) A375 cells. For SOX6 up-regulated genes, which was illustrated with OE vs. NC and WT vs. KO, revealed significant enrichment in glycolipid metabolic processes and cell cycle dysregulation, such as, “Gluconeogenesis”, “glycolytis process”, “long-chain fatty acid transport” and “positive regulation of cell cycle” (Fig. 5A and B). While overlapped down-regulated pathways were enriched in skin development, including “extracellular matrix organization”, which was detected with down-regulated in both OE vs. NC and WT vs. KO. Significant “regulation of leukocyte activation” associated genes were also down-regulated consisting with immune cell exclusion in microsatellite metastatic lesions (Fig. 5C). Conversely, metabolic validation confirmed SOX6-mediated upregulation of fatty acid transporters (FABP5, SLC2A1, SLC27A6) and lipid metabolism and transport pathways, such as, “GOBP_LIPID_IMPORT_TO_CELL” and “GOMF_FATTY_ACID_ TRANSMEMBRANE_TRANSPORTER_ACTIVITY” other than “GOBP_ POSITIVE_RETGULATION_OF_FATTY_ACID_OXIDATIONL” in GSEA (Fig. 5D; S5A).
Fig. 5
SOX6 overexpression promotes cancer cell invasion by fatty acid transport disrupt. (A) Transcriptome sequencing of SOX6_OE and SOX6_KO A375 cells, intersection between WT vs. KO and OE vs. NC, both up-regulated and down-regulated genes were selected for further analysis. (B) GO analysis of up-regulated and down-regulated genes following SOX6 overexpression. Biological processes were selected based on the count of gene number (gene count > 10) and P value (P value < 1 × 10− 2). (C) Heatmap was used for illustrated key pathways enriched in up-regulated and down-regulated genes after SOX6 dysregulation. (D) Specific up-regulation of “GOBP_LIPID_IMPORT_TO_CELL” process in cells overexpressing SOX6. (E) Specific up-regulation of long-chain fatty acid CoA biosynthesis processes in cells overexpressing SOX6. (F) Non-targeted fatty acid metabolomics examination of the fatty acid content in cells after SOX6 overexpression, demonstrating an increase in various fatty acids, particularly phospholipid components. (G) Detection of specific up-regulation of CPT1 in microsatellite lesions in ST-seq. (H) Detection of the key fatty acid transport gene ACSL3 in microsatellite lesions from clinical samples of different patients. Elevated ACSL3 expression were detected in microsatellite lesions (n = 6). Scale bars = 100 μm
To explore the oncogenic mechanisms after SOX6 overexpression in AM microsatellite lesions, we integrated the highly expressed genes from the spatial transcriptomics and SOX6 upregulated genes. The results demonstrated significant activation of glycolytic (GPI, PGM1, PGAM1, ALDOA, HK2) and lipogenic pathways [27] with concomitant NADPH/acetyl-CoA accumulation enhancing long-chain fatty acyl-CoA biosynthesis (Fig. 5E; S5B; S5C; S5D)). These results collectively indicate that SOX6 overexpression drives glycolipid metabolic dysregulation within microsatellite microenvironments.
To validate these findings, we performed untargeted fatty acid metabolomic profiling to assess fatty acid composition following SOX6 overexpression. While hypoxia universally elevated fatty acid levels, SOX6 overexpression specifically increased the phosphatidylcholine/phosphatidylethanolamine (PC/PE) ratio while reducing lysophosphatidylcholine (LPC) content (Fig. 5F; S5F; S5G). Moreover, ACSL3 and SLC27A5 were significantly up-regulated in microsatellites, indicating fatty acid transport disorder, which induces metabolic reprogramming leading to intracellular lipid accumulation (Fig. S5H; S5I). CPT1, which was also up-regulated in microsatellites, enhances long-chain fatty acid β-oxidation and generates metabolic precursors for phosphatidylcholine (PC) biosynthesis (Fig. 5G) [31]. Moreover, this specific up-regulation of CPT1 was found to be mainly driven by C1/C2 melanoma cells in AM (Fig. S5J). PC elevation combined with LPC decrease may promote tumor invasion by activating ECM-receptor interactions and HIPPO-YAP signaling pathway, which were detected in enriched KEGG pathway of ST_high and SOX6_OE UPPER genes (Fig. S5E) [32,33,34]. We observed upregulation of acyl-CoA synthetase long-chain family member 3 (ACSL3), a key fatty acid transport regulator, in microsatellite lesions across multiple patient specimens (Fig. 5H). Notably, ACSL3 upregulation was also detected in lung metastases generated by tail vein injection of SOX6_OE A375 cells, as confirmed by immunohistochemistry (Fig. S5K). Collectively, our results indicate that SOX6 overexpression promotes the glycolytic process and increases the content of long-chain phospholipids such as PE/PC by disrupting fatty acid transport and fatty acid metabolism to promote tumor invasion in C1/C2 melanoma cells.
Comments (0)