
Remember me
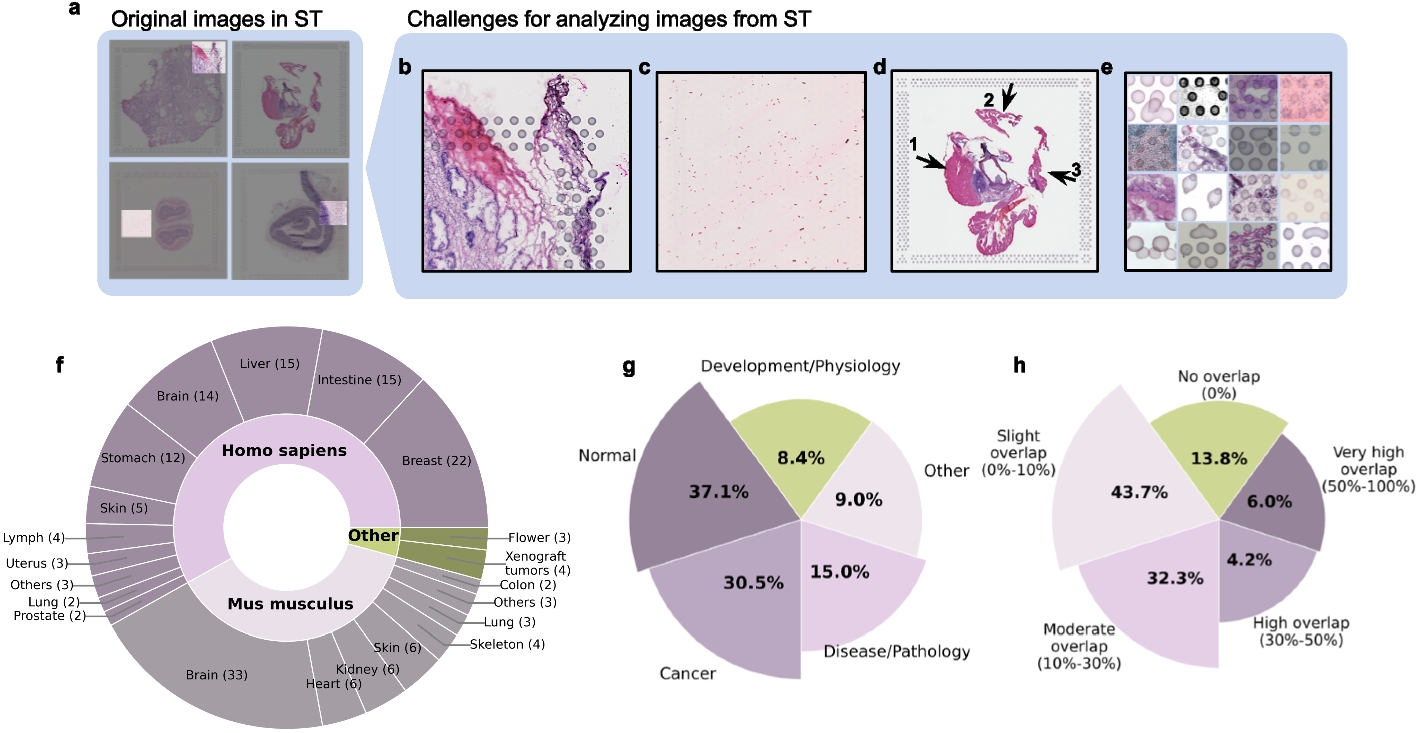


We analysed 18,764 5’UTRs annotated by the MANE project (v1 MANE Select transcripts) [27]. Of note, 298 (1.6%) MANE Select transcripts do not have an annotated 5’UTR and were excluded. We calculated the overall length of each 5’UTR as well as the position of uAUGs, and introns. The length of 5’ UTRs varies widely between genes, ranging from 1-3,561bp. The number of uAUGs ranges from 0-64 per gene, with 42.5% of 5’UTRs having at least one uAUG (Fig. 1B). We further classified these uAUGs by effect, finding that 34.4%, 15.0%, and 5.0% of 5’UTRs contain at least one uORF, oORF, and start-stop element, respectively (Fig. 1B).
In addition to annotating ‘predicted uORFs’ as all occurrences of canonical AUG triplets with an in-frame stop codon in each 5’UTR, we used a set of 5,052 functionally validated uORFs detected through ribosome profiling of six cell types and five tissues (‘Ribo-Seq uORFs’) [18]. 1,430 (28.3%) of the predicted uORFs are detected as translated in the Ribo-Seq uORFs dataset (Additional file 1: Fig. S1). In addition, the Ribo-Seq uORF set contains 2,288 additional uORFs that start at non-canonical (non-AUG) start-codons (45.3% of the Ribo-Seq uORFs). Overall, 20.9% of 5’UTRs contain one or more Ribo-Seq uORFs (range 1-11).
Genes intolerant to loss of function have longer and more complex 5’UTRsTo investigate how 5’UTRs vary by gene sensitivity to decreases in dosage we used LOEUF scores to bin genes into deciles of intolerance to LoF. The lowest deciles represent the genes most intolerant to LoF and the higher deciles represent those most tolerant [25]. We assessed 5’UTR features across LOEUF deciles. For statistical tests, we compared the lowest and highest LOEUF quintiles.
5’UTR length increases with decreased tolerance to LoF (Fig. 2A), with the 5’UTRs of genes in the lowest LOEUF quintile being significantly longer than those in the highest LOEUF quintile (mean length 269 bp vs 162 bp; Wilcoxon P<1x10-15). In other words, genes that are intolerant to LoF have significantly longer 5’UTRs. Given that LOEUF is correlated with CDS length, with shorter genes having less confident LOEUF estimates, we repeated this analysis after removing genes within the bottom 10% of CDS length. Our results remained significant (Additional file 1: Fig. S2A; Wilcoxon P<1x10-15). Further, we find that the proportion of the total mRNA that is annotated as 5’UTR is significantly greater for genes in the lowest LOEUF quintile compared to the highest quintile (Additional file 1: Fig. S2B; Wilcoxon Rank Sum, P<1x10-15) indicating that LoF intolerant genes have longer 5’UTRs even after accounting for the total length of the mRNA.
Fig. 2
Genes intolerant to LoF have longer and more complex 5’UTRs. A 5’UTRs increase in length with decreasing tolerance to LoF (Wilcoxon P<1x10-15). The average 5’UTR length across all genes (202 bp) is shown by a dotted line. The y-axis was truncated at 1,500 bp (39 genes had 5’UTRs >1,500 bp). B The 5’UTRs of genes most intolerant to LoF have lower minimum free energy (MFE) scores, representing a higher propensity to fold and create structured mRNAs (Wilcoxon P<1x10-15). The average MFE across all 5’UTRs is shown as a dotted line (-78.8). The y-axis was truncated at -1,000 (6 genes had MFE <-1000). C The 5’UTRs of genes most intolerant to LoF are more conserved. Average PhyloP scores are plotted for 5’UTRs, uORF start codons, uORF stop codons and start-stops. The dotted line denotes PhyloP=2. D Genes most intolerant to LoF are more likely to have uORFs (Chi-square P<1x10-15) and start-stops (Chi-square P=8.5x10-05) than genes most tolerant to LoF. The average numbers of each uAUG type across all 5’UTRs are shown by dotted lines. uORF: upstream open reading frame; oORF; overlapping open reading frame. E Genes most intolerant to LoF were significantly more likely to have multiple associated CAGE peaks when compared to genes most tolerant to LoF (CAGE peak >1, 91.9% vs 72.4%, Chi-square P<1x10-15; CAGE peak ≥6, 44.6% vs 16.3%, Chi-square P<1x10-15). F Whilst Ribo-seq uORFs in genes intolerant to LoF appear to more frequently have canonical start-codons, this difference is not statistically significant (Chi-square P=0.18). All statistical tests compare the lowest and highest two LOEUF deciles
Secondary structures within 5’UTRs are thought to cause inefficient ribosomal scanning [28]. The propensity of a sequence to form RNA secondary structures can be predicted from high GC content and low minimum free energy (MFE) of predicted secondary RNA structures [2, 29]. We used RNAfold [30] to compute the MFE prediction per 5’UTR. The most LoF intolerant genes had lower MFE (Fig. 2B: mean MFE=-115 vs -55, Wilcoxon P<1x10-15) and a higher GC content (Additional file 1: Fig. S3A; mean=67.3% vs 59.9%; Wilcoxon P<1x10-15) than LoF tolerant genes, indicating a higher likelihood for these 5’UTRs to be structured. To demonstrate that this greater propensity to create secondary structures is over and above what would be expected given the increased length of LoF intolerant 5’UTRs (given that longer sequences have a greater propensity to create secondary structures), we repeated the analysis only on 5’UTRs between 100-300 bp in length. The results for both MFE and GC content remained significant (both Wilcoxon P<1x10-15). These results suggest that genes that are intolerant to LoF are more likely to have stable secondary structures within their 5’UTRs.
The 5’UTRs of LoF intolerant genes are more highly conserved than LoF tolerant genes, shown by significantly higher PhyloP scores [31] (Fig. 2C; T-test P<1x10-15). This is even more pronounced when looking specifically at start and stop codons of predicted uORFs and start-stop elements (Fig. 2C; T-test all P<1x10-15). We saw a similar pattern with Combined Annotation-Dependant Depletion (CADD) scores [32] of variant deleteriousness for all possible single nucleotide substitutions at each position, with CADD scores increasing with decreased LoF tolerance (Additional file 1: Fig. S3C).
We next assessed the proportion of genes in each LOEUF decile with different categories of uAUGs. Genes most intolerant to LoF more frequently contain uORFs and start-stops than LoF tolerant genes (Fig. 2D; 46.2% vs 27.8%; P<1x10-15, and 6.8% vs 4.5%; P=8.5x10-05 for uORFs and start-stops respectively). This is true both using predicted uORFs and the uORFs detected by Ribo-Seq (Additional file 1: Fig. S4A) and remains true when correcting for different gene expression levels which can impact detection of uORFs in Ribo-seq data (Additional file 1: Fig. S5). However, we would expect there to be more uAUGs in these genes as they have longer 5’UTRs. To account for this difference in 5’UTR length across deciles, we computed the number of uAUGs per base pair (bp). The 5’UTRs of the most LoF intolerant genes have significantly fewer uAUGs per bp compared to the most tolerant genes (Additional file 1: Fig. S3D; mean=0.009 uAUG per bp vs 0.013 uAUG per bp; Chi-square P<1x10-15), suggesting that uAUGs are selectively depleted from these genes. To ensure that an overall depletion of uAUGs across 5’UTRs is not confounded by sequence composition (i.e. differences in GC content) we shuffled all MANE 5’UTR sequences 1000 times while maintaining di-nucleotide composition. AUGs were significantly more depleted than would be expected by chance (Additional file 1: Fig. S6). Despite this overall depletion, 52.1% of LoF intolerant genes (bottom quintile of LOEUF) contain at least one uAUG, suggesting that they may play an important role in translational regulation of these genes.
To determine whether the likelihood of uORF translation, and hence strength of repression of downstream CDS translation, differed between LOEUF deciles we compared the start contexts of predicted uORFs to a dataset of experimentally measured translational efficiencies (TE), quantified across a range of cell lines [6]. We saw no significant difference in TE of uAUGs across deciles (Additional file 1: Fig. S4B, Wilcoxon P=0.6), nor a significant enrichment of canonical over non-canonical start site usage of the Ribo-Seq uORFs (Fig. 2F; Chi-square, P=0.18).
Whilst we have used the MANE Select transcript set to limit our above analysis to a single, representative transcript per gene, alternative transcription start site (TSS) usage is a major contributor to transcript isoform diversity and gene regulation [33]. Cap Analysis of Gene Expression (CAGE) tags the 5’ ends of mRNA transcripts, allowing us to analyse alternative TSS usage. To observe the diversity of 5’UTRs across the LOEUF spectrum, we used CAGE data from the FANTOM consortium [34]. Genes most intolerant to LoF were significantly more likely to have multiple associated CAGE peaks when compared to genes most tolerant to LoF (Fig. 2E; CAGE peak >1, 91.9% vs 72.4%, Chi-square P<1x10-15; CAGE peak ≥6, 44.6% vs 16.3%, Chi-square P<1x10-15). As this analysis may be confounded by gene expression levels, with more highly expressed genes having more associated CAGE peaks, we repeated this analysis splitting genes into four quartiles of mean expression across tissues in GTEx. The result remained significant in all four quartiles (Additional file 1: Fig. S7; all Chi-square, P<8x10-11). Assessing alternative splicing possibilities, we found no significant difference in the proportion of genes that have 5’UTR introns across LEOUF deciles (Additional file 1: Fig. S3B, Chi-square P=0.19).
Finally, we hypothesised that the uORFs in LoF intolerant genes might be optimised to promote efficient uORF translation and re-initiation at the CDS start-codon. We assessed codon optimality (tAI scores) of the Ribo-Seq uORFs, but found no significant differences between deciles (Additional file 1: Fig. S8A; Wilcoxon P=0.17). We observed a very small, but significant difference in average uORF length across deciles (means 52.5 bp vs 59.1 bp, Wilcoxon P=4.9x10-06), but only when considering the predicted uORF and not the Ribo-Seq set (Additional file 1: Fig. S8B, 8C; Wilcoxon P=0.9). We also observed that the stop codons of the uORFs closest to the CDS start are significantly further upstream of the CDS start in more LoF intolerant genes (Additional file 1: Fig. S8D; means 99 bp vs 77 bp, Wilcoxon P=1.3x10-04). In other words, these genes have a greater potential re-initiation distance.
Translational regulation through 5’UTRs is important for genes involved in diseaseGiven the increased complexity of 5’UTRs observed in LoF intolerant genes, we were interested to see whether these results were relevant to 5’UTRs of genes where disruption of tight regulatory control may lead to disease. We investigated 5’UTR features in genes within which predicted LoF variants have been reported to cause developmental disorders (DD) and cancer, as well as a wider set of dosage sensitive (DS) genes [35,36,37]. For DD genes, we compared dominant and recessive genes, given the former are more likely to be highly dosage sensitive. For cancer, we analysed tumour suppressor genes (TSGs) and oncogenes separately (Onc). Finally, for DS genes we compared haploinsufficient (HS) and triplosensitive (TS) genes. For all statistical tests we compared the disease gene group against all MANE Select 5’UTRs with that specific disease group removed.
Whilst 5’UTRs average 202 bp in length, disease gene 5’UTRs are significantly longer (Fig. 3A; DD dominant: 369 bp, Wilcoxon P<1x10-15; Onc: 260 bp, Wilcoxon P=1.5x10-05; TSG: 254 bp, Wilcoxon P=2.9x10-04; HS: 279 bp, Wilcoxon P<1x10-15; TS: 253 bp, Wilcoxon P<1x10-15). A significantly higher number of disease gene 5’UTRs contain uORFs than the average of 34.4% across all genes (Fig. 3C; DD dominant: 57.9%, Chi-square P<1x10-15; TSG=49.4%, Chi-square P=6.5x10-05; HS=45.7%, Chi-square P<1x10-15; TS=40.7%, Chi-square P=1.4x10-07), although the difference is not-significant for the oncogene gene set (43.1%, Chi-square P=0.07). Start-stop elements are only significantly enriched in HS genes (7.1% vs 5.0%; Chi-square P=3.1x10-08), however, given the small number of genes that contain start-stops, we are likely underpowered to detect a significant enrichment in our smaller gene sets.
Fig. 3
Comparison of 5’UTRs across disease genes sets. A The 5’UTRs of disease genes are significantly longer (Wilcoxon: DD dominant: P<1x10-15; Onc: P=1.5x10-05; TSG:P=2.9x10-04; HS: P<1x10-15; TS: P<1x10-15) with the exception of DD recessive genes which are significantly shorter (Wilcoxon P=2.7x10-08), when compared to the average across all genes. The median 5’UTR length for all genes (136 bp) is shown by the dotted black line. The x-axis was truncated at 2,000 bp (22 genes had 5’UTRs >2,000 bp). B Disease gene 5’UTRs are significantly more conserved (T-test: DD dominant: P<1x10-15; Onc: P=9x10-06; TSG: P=4.3x10-08; HS: P<1x10-15; TS: P<1x10-15) except DD recessive genes which are significantly less conserved (T-test: P=4.9x10-08), compared to all genes. The dotted black line is the median PhyloP score for all genes (0.28). C Disease genes significantly more often contain uORFs (Chi-square: DD dominant: 57.9%, P<1x10-15; TSG=49.4%, P=6.5x10-05; HS=45.7%, P<1x10-15; TS=40.7%, P=1.4x10-07), when compared to all 5’UTRs. Start-stops are only significantly enriched in HS genes (P=3.1x10-08). The dotted lines mark the percentage of all genes with each uAUG type
Disease gene 5’UTRs are also significantly more conserved when compared to all genes (Fig. 3B; DD dominant: T-test P<1x10-15; Onc: T-test P=9x10-06; TSG: T-test P=4.3x10-08; HS: T-test P<1x10-15; TS: T-test P<1x10-15). We did not observe a significant difference in the number of 5’UTR introns between disease gene sets and all genes (Additional file 1: Fig. S9; DD dominant: Chi-square P=0.07; Onc: Chi-square P=0.09, TSG: Chi-square P=0.15; HS: Chi-square P=0.18; TS Chi-square P=0.39).
We observed a marked distinction between DD dominant and recessive gene 5’UTRs. When compared to the average across all genes, the 5’UTRs of DD recessive genes were significantly shorter (Fig. 3A; mean=169 bp, Wilcoxon P=2.7x10-08), have significantly fewer 5’UTR introns (Additional file 1: Fig. S9; Chi-square P=4.7x10-06), are significantly less conserved (Fig. 3B; PhyloP, T-test P=4.9x10-08), and also have fewer uORFs and start-stops (Fig. 3C; Chi-square P=2.7x10-06 (uORFs); Chi-square P=1.3x10-04 (start-stops)). The lower complexity of the 5’UTRs of this recessive gene set likely reflects their insensitivity to changes in dosage. The observation that these 5’UTRs are significantly different to the all gene average likely reflects the fact that the all gene set contains many genes that are sensitive to dosage changes. To account for this, we tested DD recessive genes against genes in the middle two LEOUF deciles; we see no significant difference in 5’UTR length (mean 169 vs 177 bp, Wilcoxon P=0.04), the number of uORFs (Chi-square P=0.66), or mean PhyloP scores (T-test P=0.1). We do still observe significantly fewer introns in DD recessive genes (Chi-square P=8.2x10-05).
Visualising 5’UTRs with VuTRHere, we have presented an overview of 5’UTRs across different gene sets, however, there is still considerable variability within each set. To support investigation of individual gene 5’UTRs, their regulatory features, and genetic variation within them, we have created an interactive web-based tool, VuTR (pronounced view TR; https://vutr.rarediseasegenomics.org/). For a query gene symbol or MANE transcript ID, VuTR displays the sequence of the 5’UTR, statistics including the length and number of uAUGs, and the distribution of both predicted and Ribo-Seq uORFs within the 5’UTR. Further, VuTR uses annotations from UTRannotator [38] to display variants in gnomAD [25] and ClinVar [39] that create uAUGs or disrupt predicted uORFs. Figure 4 shows the output for NF1.
Fig. 4
VuTR: an interactive web-based tool. A A screenshot from VuTR showing the NF1 gene. The top section displays summary gene details and links to other tools and databases. The following section, titled ‘5’UTR Architecture’, shows the gene’s native 5’UTR exon structure, predicted uAUG elements, and Ribo-Seq uORFs. NF1 features two predicted uORFs, both with a strong Kozak consensus strength (shown by the yellow colour), the longer 45 bp uORF is also found in the Ribo-Seq dataset. Variants observed in gnomAD and ClinVar are displayed in separate tracks. Each variant that creates a uORF or disrupts a predicted uORF is shown on a separate row. Here, a variant that disrupts the start of the longer predicted uORF, which is also found in the Ribo-Seq data (uAUG-lost; 17-31094995-T-G) is observed in gnomAD. Four ClinVar variants create out-of-frame ORFs (oORFs) by either disrupting the stop codon of the two native uORFs (uSTOP-lost; 17-31095037-A-T, 17-31095037-A-G, 17-31095038-G-C) or by creating a new oORF through a uAUG-gained variant (17-31095038-G-A). B An example of a popup that appears when a uORF / oORF is selected, giving context specific details regarding its sequence, Kozak consensus strength, and a histogram of how its predicted translational efficiency [(6)] compares to all other uORFs / oORFs within MANE 5’ UTR sequences. C An example of a popup that appears when a ClinVar variant is selected. This example shows the uAUG-creating variant, 17-31095038-G-A. The popup displays the variant details, information from ClinVar, and the variant annotation from UTRannotator [38]. uORF: upstream open reading frame
Comments (0)