
Remember me
Cell lines CTLL-2 (American Type Culture Collection (ATCC)), Expi293F (Gibco) and HEK-Blue IL-12 (Invivogen) were cultured according to vendor instructions. MC38 (Kerafast, cat. no. ENH204-FP), MC38-ZsGreen (developed in the lab as described previously40) and B16F10 (ATCC, cat. no. CRL-6475) cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 100 units ml−1 of penicillin and 100 μg ml−1 of streptomycin. The 4T1-GFP-Luc (4T1-Luc) cells were generated by transduction of the 4T1 cell line (ATCC, cat. no. CRL-2539) with pGreenFire lentiviral vector (System Biosciences) as described previously41. The 4T1-Luc cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% FBS, 100 units ml−1 of penicillin and 100 μg ml−1 of streptomycin. All cells were maintained at 37 °C and 5% CO2 and all were confirmed to be negative for Mycoplasma spp.
MiceAll animal studies and procedures were carried out following federal, state and local guidelines under an institutional animal care and use committee-approved animal protocol by the Committee of Animal Care at MIT. For all studies involving animals, female BALB/c (Jackson Laboratory, cat. no. 000651), C57BL/6J (Jackson Laboratory, cat. no. 000664) and Batf3−/− (Jackson Laboratory, cat. no. 013755) mice aged 6–8 weeks were purchased and maintained in the animal facility at Massachusetts Institute of Technology (MIT). Specific strains associated with experiments are listed in the figure captions. All mice were housed in a specific pathogen-free facility, and fed normal chow and water freely under standard animal facility conditions (12 h light:dark cycle). No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications10,11,12,42,43. Data collection and analysis were not performed blind to the conditions of the experiments.
Cloning and protein purificationGene blocks (gBlock, IDT) encoding for the light and heavy chain variable regions of anti-CD45 (clone 30-F11 (ref. 44)) or untargeted isotype control (anti-FITC, clone 4-4-20) were cloned into a mouse κ light chain and IgG2c backbone with LALA-PG20 mutations, respectively, in the mammalian expression vector gWiz (Genlantis). For αCD45-IL-15, murine IL-15/IL-15Rαsushi, as described previously11, was then cloned at the C terminus of the anti-CD45 (or anti-FITC control) heavy chain. To generate αCD45-IL-12, murine single chain IL-12 (scIL-12), as described previously11, was cloned at the amino terminus of the anti-CD45 (or anti-FITC control) heavy chain. For both IL-15 and IL-12 immunocytokines, a (Gly4Ser)3 linker was used between the cytokine and antibody. To generate human αCD45-IL-15, gBlocks encoding for the light and heavy chain variable regions of anti-human CD45 (clone BC8) were cloned into a human κ light chain and human IgG1 backbone with LALA-PG mutations, respectively. Human IL-15/IL-15Rαsushi, as described previously11, was then cloned at the C terminus of the anti-CD45 heavy chain.
A gBlock encoding for the extracellular domain of mouse CD45RO (used for ELISAs; sequence obtained from UniProt) was cloned into gWiz with a His6 tag. Plasmid sequences confirmed by Sanger sequencing (Quintara Biosciences) were transformed into Stellar Competent Cells (Takara Bio) and purified using the NucleoBond Xtra Midi endotoxin-free midi-prep kit (Takara Bio). For immunocytokines, an equal mass of heavy and light chain plasmids was transfected into Expi293F cells (Gibco) per the manufacturer’s instructions and harvested 6 d after transfection. All immunocytokines were purified using rProteinA Sepharose Fast Flow resin (Cytiva Life Sciences) and validated for size by sodium dodecylsulfate–polyacrylamide gel electrophoresis. His-tagged CD45RO was purified by TALON affinity resin (Takara) according to the manufacturer’s instructions. Purified proteins were confirmed to be endotoxin free (<0.1 endotoxin units (EU) per dose) using the Endosafe Nexgen-PTS system (Charles River). Purified proteins were flash frozen in liquid nitrogen and stored at −80 °C until use.
ELISA and bioactivity assaysFor ELISA assays, Maxisorp 96-well flat-bottomed plates (Thermo Fisher Scientific) were coated with recombinant CD45RO at 0.2 µg ml−1 in phosphate-buffered saline (PBS; Corning) overnight at 4 °C. Subsequent washes were performed with PBST (PBS supplemented with 0.05% v:v Tween-20 (Millipore-Sigma)). Blocking was performed in PBSTA (PBS supplemented with 1% w:v bovine serum albumin (BSA; Sigma-Aldrich) and 0.05% v:v Tween-20) overnight. Immunocytokines were diluted to relevant concentrations in PBSTA and detected via a horseradish peroxidase-conjugated anti-mouse IgG secondarily diluted 1:3,000 (Abcam). One-Step Ultra TMB-ELISA substrate solution (Thermo Fisher Scientific) was added to develop for 5 min and quenched with 2 N sulfuric acid (VWR). Absorbance at 450 nm (A450) with reference at 570 nm was measured on an Infinite M200 microplate reader (Tecan).
For IL-15 bioactivity, 10,000 CTLL-2 cells were seeded in a 96-well, U-bottomed plate in incomplete medium per the manufacturer’s instructions with stated IL-15 immunocytokine dilutions for 48 h at 37 °C. Proliferation was measured via CellTiter-Glo 2.0 Assay (Promega) following the manufacturer’s instructions. Luminescence was measured on a microplate reader (Tecan) with an integration time of 0.25 s. For the CD45 pre-blockade experiment, cells were incubated with 500 nM αCD45 (clone 30F11) for 4 h before the addition of IL-15 immunocytokine. IL-12 bioactivity was measured with HEK-Blue IL-12 reporter cells according to the manufacturer’s instructions (Invivogen).
Primary CD8+ T cell preparationSpleens from 6- to 8-week-old C57BL/6J female mice were harvested and processed into single-cell suspensions. CD8+ T cells were isolated using the EasySep Mouse CD8+ T cell isolation kit (StemCell Technologies) and resuspended at a concentration of 106 cells ml−1 in complete RPMI supplemented with 1× sodium pyruvate (Thermo Fisher Scientific), 1× nonessential amino acids (Thermo Fisher Scientific) and 1× 2-mercaptoethanol (Thermo Fisher Scientific). Medium was additionally supplemented with 10 ng ml−1 of murine IL-2 (BioLegend) before resuspension and subsequent passaging. Isolated CD8+ T cells were activated for 48 h on a 6-well, nontissue culture-treated plate that was precoated with 0.5 µg ml−1 of anti-CD3 (BioXCell, clone 2C11) and 5 µg ml−1 of anti-CD28 (BioXCell, clone 37.51) overnight at 4 °C. The plate was washed twice before activation. After activation, T cells were cultured for 48 h before use in downstream experiments.
For human CD8+ T cell work, CD8+ T cells isolated from PBMCs were purchased from StemCell Technologies. Isolated cells were activated with αCD3/αCD28 Dynabeads (Thermo Fisher Scientific) at a 1:1 ratio in complete T cell medium (described above) supplemented with 10 ng ml−1 of human IL-2 for 72 h. Cells were then rested for at least 48 h before use in downstream assays.
Fluorescence-quenching internalization assayInternalization assays were performed and analyzed as described previously45. Briefly, immunocytokines were conjugated with Alexa Fluor-488 (AF488) using N-hydroxysuccinimide (NHS) ester chemistry (Invitrogen). Free dye was removed by Zeba spin desalting column purification (Thermo Fisher Scientific). Primary CD8 T cells, 100,000, were seeded in a 96-well plate and incubated with AF488-labeled immunocytokines at 10 µg ml−1 staggered at desired time points. Wells were then split such that one set was incubated with 25 µg ml−1 of anti-AF488-quenching antibody (Thermo Fisher Scientific, cat. no. A-11094) for 25 min. For human experiments, anti-human CD45 (clone MEM-28) conjugated to AF488 (Thermo Fisher Scientific) was used. IL15/IL15R was purchased from MedchemExpress. Viability was assessed via DAPI staining. The AF488 signal was measured using a BD LSR Fortessa and FACSDiva software and data were analyzed in FlowJo.
Analysis of STAT phosphorylation by flow cytometryFor in vitro STAT5 experiments, primary CD8+ T cells cultured as described above were starved of IL-2 for 24 h and seeded into 96-well plates at 100,000 cells per well. IL-15 immunocytokines were added at 10 µg ml−1 for 1 h and subsequently washed with incomplete T cell medium (no IL-2) twice before resting for 24 h. IgG-IL-15 added at the same molar concentration (without washing) was used as a continuous control. Cells were immediately fixed in medium with equal volumes of BD Phosflow Fixation Buffer I prewarmed to 37 °C for 10 min. When required, cells were stained with Zombie Aqua viability stain for 5 min in PBS (1:1,000 dilution) before fixation. Cells were permeabilized for 30 min on ice with BD Phosflow Perm Buffer III that had been pre-chilled to −20 °C. Staining with anti-pSTAT5 antibodies (BD, clone 47) conjugated to AF647 or phycoerythrin (PE) was carried out at room temperature for 1 h. STAT4 experiments were performed identically, but used complete medium supplemented with IL-2 and immunocytokine incubation was performed at 2 µg ml−1 for 20 min. The pSTAT4 signal was detected with anti-pSTAT4 (BD, clone 38). For trans-signaling experiments, immunocytokines were conjugated with AF647 using NHS ester chemistry. Preloaded cells were labeled with CFSE per the manufacturer’s instructions. For cis-signaling experiments, CD8+ T cells were diluted with HEK293F (CD45−IL-15R−) at the stated ratios before incubation with αCD45-IL-15 at the stated concentrations. The total number of cells in the well was maintained at a constant level.
Measurement of STAT5 levels in vivo was carried out as previously described46. Briefly, TDLNs were processed into single-cell suspensions directly in BD Fixation Buffer I and samples were incubated at 37 °C for 10 min. Downstream permeabilization and staining were performed as described above. In all cases, the pSTAT signal was measured using a BD LSR Fortessa and FACSDiva software and data were analyzed in FlowJo.
Tumor inoculation and treatment preparationFor all single-tumor experiments, mice aged 6–8 weeks were injected subcutaneously (s.c.) in the shaved right flank with 106 tumor cells (MC38, MC38-ZsGreen or B16F10) in a volume of 50 μl of PBS. For two-tumor experiments, the contralateral tumor was inoculated on the left flank 3 d after the primary tumor, as stated in the study schematics. Before treatment, mice were randomized to ensure equal mean initial tumor size across groups. Immunocytokines were prepared at their stated doses (1 μg for IL-12 immunocytokines and 10 μg for IL-15 immunocytokines, where the mass indicated is the mass of the entire fusion protein) and dosed i.t. in 20 μl of PBS unless otherwise stated. Doses were informed by our biodistribution experiments as well as previous intratumoral cytokine work from our lab10. Peritumoral administration was performed in 50 μl of PBS injected s.c. at the tail base. Intraperitoneal administration was performed in 100 μl of PBS. The tumor area was calculated as the product of tumor length and width. For single-tumor studies, mice were euthanized when the tumor area exceeded 100 mm2; for two-tumor studies, mice were euthanized when cumulative tumor area exceeded 200 mm2. The maximal tumor burden was not exceeded on any study in this work. Immune memory rechallenge experiments were carried out 100 d after initial challenge with 105 tumor cells on the left flank. Age-matched naive mice were used as controls for these studies. For the lung metastasis model used in Extended Data Fig. 8, 2 × 105 B16F10 cells in 100 μl of PBS were inoculated retro-orbitally on the same day as the standard 106 tumor-cell flank inoculation.
4T1 orthotopic inoculation and metastasisFor 4T1-Luc experiments, 0.5 × 106 4T1-Luc cells were inoculated into the fourth mammary fat pad of BALB/c mice. Then 12 d after the start of treatment, mice were anesthetized with isoflurane and provided preoperative, subcutaneous, sustained-release buprenorphine (1 mg kg−1; ZooPharm) and meloxicam (5 mg kg−1). The primary tumor was surgically removed and the wound was closed with surgical clips. Meloxicam (5 mg kg−1) was dosed every 24 h for 3 d postoperatively. Bioluminescence-imaging, 4T1-Luc tumor–bearing mice were monitored postoperatively for development of metastases using bioluminescence imaging beginning 1 week after surgical resection of the primary tumor. Animals were injected i.p. with 150 mg kg−1 of sterile filtered d-luciferin (PerkinElmer) in 200 ml of sterile PBS. Animals were imaged 15 min after the d-luciferin injection using the IVIS Spectrum Imagining System 100 (Xenogen).
Tissue processing for flow cytometryB16F10 or MC38 tumors were harvested, weighed and subsequently minced using dissection scissors in gentleMACS mouse tumor dissociation buffer (Miltenyi) prepared per the manufacturer’s instructions. As noted in the Miltenyi protocol, enzyme R was reduced to 20% of the stated amount to preserve surface epitope integrity. Minced tumors were processed on a gentleMACS Octo-dissociator with heaters (Miltenyi) using program mTDK_1 for B16F10 and mTDK_2 for MC38. Dissociated tumors were then filtered through a 70-μm strainer and 25 mg of tumor was plated for downstream staining. TDLNs were harvested, weighed and subsequently dissociated and filtered through a 5-ml round-bottomed tube with cell-strainer cap (Falcon) using the blunt rubber end of a 1-ml syringe plunger (Falcon). Then, 5 mg of TDLNs was used for downstream staining. Blood was collected by submandibular bleeding into MiniCollect K2-EDTA tubes (Greiner) and red blood cells were lysed using ACK Lysis Buffer (Gibco). When intracellular cytokine staining was performed, as in Fig. 5, samples were resuspended and plated in complete RPMI supplemented with 1× sodium pyruvate, 1× nonessential amino acids, 1× 2-mercaptoethanol and 1× brefeldin A (BioLegend) and allowed to incubate at 37 °C for 3 h before staining. Precision counting beads (50 μl, BioLegend) were added after initial resuspension and used for downstream data analysis. Viability was assessed with Zombie UV or Zombie NIR dyes (BioLegend, 1:1,000) in PBS for 20 min at room temperature. Subsequent washes and surface staining were performed in PBS supplemented with 1% BSA and 2 mM EDTA (Thermo Fisher Scientific). Samples were resuspended in Mouse Fc block Plus (BioLegend) before surface staining for 15 min. The antibodies against surface targets used are the following: CD3 (BD, cat. no. 612803, clone 17A3, 1:100), CD4 (BD, cat. no. 612923, clone GK1.5, 1:100), CD8 (BioLegend, cat. no. 100723, clone 53-6.7, 1:200), CD19 (BioLegend, cat. no. 115543, clone 6D5, 1:100), CD24 (BioLegend, cat. no. 101822, clone M1/69, 1:100), CD25 (BioLegend, cat. no. 102034, clone PC61, 1:100), CD44 (BD, cat. no. 612799, clone IM7, 1:100), CD45.2 (BD, clone: 104, 1:100), NK1.1 (BioLegend, cat. no. 108741, clone PK136, 1:100), MHC-II (BioLegend, cat. no. 107628, clone M5/114.15.2, 1:100), Ly6C (BioLegend, cat. no. 128036, clone HK1.4, 1:100), F4/80 (BioLegend, cat. no. 123147, clone BM8, 1:100), PD-1 (BioLegend, cat. no. 135220, clone 29F.1A12, 1:100) and TIM3 (BioLegend, cat. no. 119725, clone RMT3-23, 1:100). P15E tetramer (MBL) staining was performed in the presence of 50 nM dasatinib at a 1:75 dilution and anti-CD8 antibody clone KT15 (Thermo Fisher Scientific) was used to minimize background signal. Dasatinib incubation was not included in the staining mixture for the RNA-seq experiment. When performing intracellular staining, cells were fixed and permeabilized using the Foxp3 transcription buffer set (eBioscience). The samples against intracellular antigens used are as follows: TCF1 (Cell Signaling Technologies, cat. no. 90511, clone C63D9, 1:250), IFNγ (BioLegend, cat. no. 505808, clone XMG1.2, 1:200) and granzyme B (BioLegend, cat. no. 396418, clone QA16A02, 1:200). Intracellular staining was performed overnight at 4 °C. Cells were collected using a BD FACSymphony A3 and FACSDiva software and data were analyzed in FlowJo.
Antibody-mediated cellular depletionImmune cell depletions were carried out with antibodies targeting CD8a (BioXCell, clone 2.43, 400 μg twice weekly), NK1.1 (BioXCell, clone PK136, 400 μg twice weekly) and CSF1R (BioXCell, clone AFS98, 300 μg every other day) as previously described11. All depletions were given i.p. in 100 μl of PBS. Depletions were initiated 1 d before treatment and carried out for 4 weeks. Depletions were carried out in C57BL/6J mice unless otherwise noted.
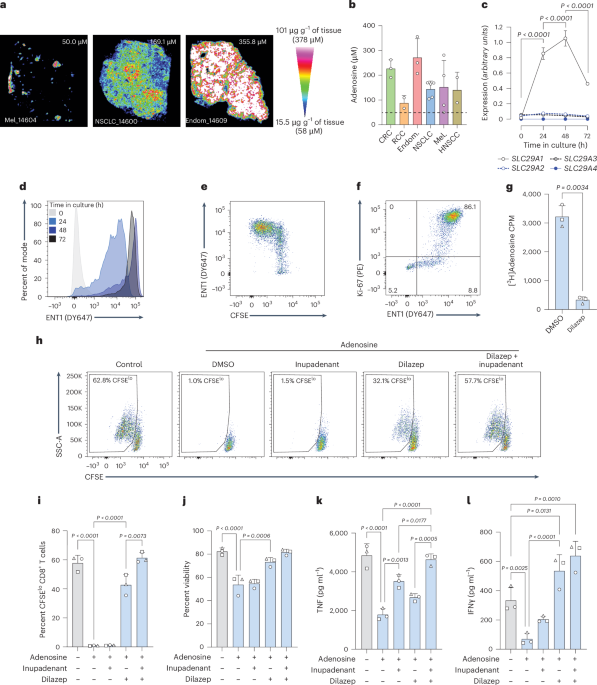
Tissue lysates for biodistribution and analyte measurementIL-15 immunocytokines were labeled with AF647 using NHS ester chemistry per the manufacturer’s instructions. Free dye was removed with Zeba desalting columns. Molar amounts of dye for each immunocytokine were matched before dosing. Tissues were excised, weighed and processed in a GentleMACS M Tube using the program Protein_01 in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific) supplemented with Halt Protease Inhibitor Mixture (Thermo Fisher Scientific). Samples were pelleted by centrifugation at 10,000g to remove debris and supernatants were pipetted on to a 384-well plate. Fluorescence was measured on a microplate reader (Tecan) with gain and z-value optimized for each tissue. Cytokine concentrations were calculated based on a standard curve prepared using known amounts of serially diluted AF647-labeled immunocytokine at the gain and z-value optimized for that given tissue. Serum mass density was taken to be 1 g ml−1 for conversion. The LEGENDplex mouse antivirus response panel (13-plex; BioLegend) was used following the vendor instructions.
FTY720 preparation and dosingFTY720 hydrochloride (Sigma-Aldrich) was stored in stock solutions at 10 mg ml−1 in dimethyl sulfoxide. Before treatment, stock solutions were diluted to a dose of 30 μg in 150 μl in PBS. In two-tumor MC38 studies, FTY720 was dosed every other day i.p. starting on day 5 after tumor inoculation.
Leukocyte fraction analysisTo compare the CD45 infiltration of mouse and human tumors, we used existing datasets from The Cancer Genome Atlas (TIMER2.0 database47) or a comprehensive syngeneic mouse database (TISMO database48). The leukocyte fraction was calculated from EPIC RNA-seq analysis as the sum of B cell, NK cell, CD8, CD4 and macrophage fractions.
Immunofluorescence stainingInguinal LNs and tumors were harvested 24 h post-injection as described in Fig. 2a, embedded in optimal cutting temperature buffer (Fischer Scientific) and fresh frozen. Then 10-μm tissue sections were post-fixed with 4% paraformaldehyde (PFA) for 10 min, followed by three washes with PBS. Sections were incubated with Fc receptor blocker (Innovex) for 30 min and blocked for 1 h with 5% goat serum and 2.5% BSA in PBS. Staining with primary antibodies was performed overnight at 4 °C in blocking buffer (LN:IgD; BioLegend, cat. no. 405705) and CD8 (Abcam, cat. no. ab217344); tumors: CD8 and F4/80 (Abcam, cat. no. ab105156). After three washes with PBS, the sections were incubated with fluorochrome-conjugated secondary antibody (Thermo Fisher Scientific, cat. no. 35551) in blocking buffer for 30 min at room temperature. After three washes with PBS, the sections were mounted on to glass slides using mounting medium (ProLong Diamond Antifade Mountant, Thermo Fisher Scientific). High magnification images were acquired using a Leica SP8 laser-scanning confocal microscope equipped with a white light laser, a 405 solid state laser line and selective emission filters. Images were collected using a ×25 water immersion lens and a ×63 oil immersion lens.
Immunohistochemistry staining of lung sectionsAnimals were euthanized and transcardially perfused with PBS before harvesting the lungs. Tissues were fixed overnight in 4% PFA at 4 °C, processed using conventional methods, embedded in paraffin and sectioned at 10 μm. Sections were then stained with hematoxylin and eosin and scanned using the Aperio Brightfield (Leica Biosystems) Slide Scanning System. The lung tissue and metastatic lesions were automatically detected via distinct pixel classifiers using QuPath v.0.4.3.
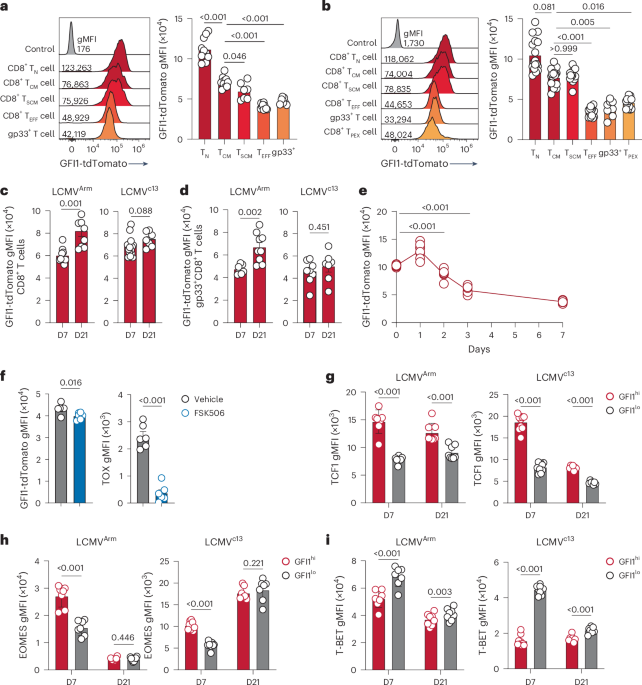
CD8+ T cell RNA-seq, mapping and analysisFor RNA-seq experiments, 2,000–40,000 live CD3+CD8+CD44+p15E+ cells processed from TDLNs were sorted using a Sony MA900. RNA extraction was performed using the QIAGEN RNEasy Micro kit per the manufacturer’s instructions. RNA libraries were prepared using the Clontech SMARTer Stranded Total RNA-Seq Kit—Pico Input Mammalian and sequenced using the Illumina NextSeq500 75-nt kit. RNA-seq reads were aligned to the mouse genome with STAR (v.2.7.9a) using ensembl GRCm39 primary assembly as the reference. Aligned reads were quantified using RSEM (v.1.3.1) with ensembl GRCm39 (release 110) transcript annotations. The resulting counts were analyzed in R using DESeq2 for differential expression analysis, fgsea for GSEA and msigDB for the gene set database. Data visualization was done with ggplot2 and ComplexHeatmap. GSEA was utilized for the correlative analysis between our RNA-seq data and the gene expression signatures from Prokhnevska et al.25. The gene expression count matrix was obtained from National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) with accession no. GSE216731. Differential gene expression analysis was performed on the LCMV Arm and TDLN groups and the genes were ranked by Wald’s test statistics. The ranked genes were compared with gene signatures from our data, specifically the upregulated genes in αCD45-Cyt versus untreated mice and the upregulated genes in untreated versus αCD45-Cyt mice. The enrichment score suggests the degree of correlation with T cells from either the LCMV Arm or TDLNs.
Statistical methodsStatistics were computed in GraphPad Prism v.9 as denoted in the figure captions. For in vitro biodistribution and flow cytometry immunophenotyping experiments, comparisons were made by two-sided Student’s t-test or one- or two-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test. Survival comparisons were made by log(rank) (Mantel–Cox) tests. Unless noted otherwise, data distributions were assumed to be normal but this was not formally tested. Differential gene expression analysis in the RNA-seq data was performed by two-sided Wald’s tests. In all RNA-seq analyses, P values are corrected by Benjamini–Hochberg to account for multiple hypothesis testing. No data/experiments were excluded unless there were technical issues with the experiment and outliers were not excluded. Exact P values are denoted in the figures. For all figures, NS is not significant (P > 0.05).
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)