
Remember me
ENT1-KO mice were generated upon request from Cyagen, using a C57BL/6J background. Specifically, a genomic region corresponding to exons 1–12 was deleted from the mouse genome through the application of CRISPR–Cas9 technology. To validate the KO, germ lines underwent downstream analysis, including DNA sequencing and protein detection using western blotting. The colony of ENT1-KO mice was established and maintained at Taconic Facilities in Denmark. Once established, the mice were transported to the animal facility at Université Libre de Bruxelles (ULB) for experimentation. For comparative purposes, 7–8-week-old WT C57BL/6J female mice were obtained from Taconic (Denmark).
Mice were housed under pathogen-free conditions at ULB facilities, following all animal guidelines set by FELASA (Federation of European Laboratory Animal Science Associations). Mice were maintained on 12 h light/dark cycles, and the housing facility was kept at 20–25 °C and 30–70% humidity. Mice were fed CRM (P) (DS801727G10R) from SDS Diets. Animal protocols underwent thorough review and received approval from the Institute of Animal Care and Use Committee at ULB (agreement LA-150055-18, project code BUC 2020-03).
Cell linesCT26, HT29, A375, JAR, MDA-MB-231 and T2 cells were obtained from ATCC (American Type Culture Collection). MCA205 and MC38 cells were acquired from Sigma-Aldrich. KPC cells (clone 6419c5) were obtained from Kerafast. Pan02 cells were obtained from the National Cancer Institute Division of Cancer Treatment and Diagnosis Tumor Repository.
ENT1-KO JAR cells were generated using CRISPR–Cas9 technology, with delivery of the ribonucleoprotein complex to the JAR cells through Lipofectamine CRISPRMAX Cas9 Transfection Reagent. The specific single guide RNA (sgRNA) was designed to target exon 5, and its sequence was GCTCAGTTCAGCAGTGACCA. After 1 week of ribonucleoprotein delivery, the cells were stained with SAHENTA-DY647 dye to identify ENT1 expression, and the negative population was isolated through single-cell sorting by FACS. The resulting clones were subsequently expanded and subjected to validation through Sanger sequencing and western blotting.
MCA205-ENT2-KO cells were generated as described above using the sgRNA GCAAGCCAGCCTTTTTGGTC and GAGCTGTCAGAAAGCGCCTT to target exons 7 and 8, respectively.
All tumor cell lines were tested regularly for mycoplasma contamination by PCR and were verified to be negative before using them for different assays and studies.
Cell cultureCell lines were cultured following the provider’s recommendations. In brief, MCA205 was cultured in DMEM high glucose (Lonza) supplemented with 10% FBS (Gibco) and 1.5% of HEPES (Lonza); MC38, KPC and A375 were cultured in DMEM (Lonza) with 10% FBS; CT26, Pan02 and JAR were cultured in RPMI (Lonza) supplemented with 10% FBS and 1% HEPES; MDA-MB-231 was cultured in L-15 medium supplemented with 10% FBS and 1% glutamine (Gibco); and HT29 was cultured with McCoy’s 5a supplemented with 10% FBS. All cell lines were cultured in culture flasks (Greiner) and incubated at 37 °C with 5% CO2. For in vivo studies, the cells were expanded and passaged three times before inoculation.
Mouse syngeneic tumor experimentsCultured tumor cells were dissociated into single cells with 0.25% trypsin (Gibco), washed with serum-free medium and counted in preparation for subcutaneous implantation. The cells were then resuspended in 100 µl of PBS. The total number of inoculated cells was specific to each model: MCA205 and KPC, 2 × 105; MC38 and CT26, 5 × 105; Pan02, 2 × 106. Tumor-bearing mice were randomized into groups with equivalent average tumor volumes when the tumors reached 60 mm3. The animals were monitored daily, checking for clinical signs of distress and recording body weight, and tumors were measured three times per week using an electronic caliper. Tumor volume was determined as volume = length × width2 × 0.5.
In some experiments, CD8+ T cell depletion was performed by injecting 250 µg of anti-CD8 monoclonal antibody (clone 53-6.7, BioXcell) intraperitoneally on days −7, −4, 0, 7, 14 and 21, with day 0 marking tumor implantation.
Tumor growth inhibition was calculated after logarithmic transformation of tumor volumes. Times were transformed to orthogonal polynomials, making the intercept of the analysis equivalent to overall tumor burden (area under the curve) during the entire experiment53. Maximal tumor size was 2,000 mm3 and was not exceeded in the study.
Human xenografts in humanized miceHuman cell line xenograft experiments were performed at TransCure BioServices (France), and all associated animal procedures were reviewed and approved by the local ethics committee (CELEAG–TCS agreement number A7418324.09DEC2021: APAFIS##30873-202112011817784 V2; 17MAR2021: APAFIS#38383-2022082413416895 V7). The experiment was carried out using the NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl immunodeficient mouse strain (NCG, from Charles River Laboratories). Mice were housed on a 12 h light/dark cycle in a housing facility maintained at 20–25 °C and 30–70% humidity and were fed a diet of Teklad Global 18% Protein Rodent Diet (2018) (Envigo, formerly Harlan). In brief, 4-week-old immunodeficient NCG mice were engrafted with cord-blood-derived human CD34+ hematopoietic stem and progenitor cells (French Blood Institute) 2 days after chemical myeloablative treatment. Then, 14 weeks after cell injection, the engraftment level was monitored with the analysis of human CD45+ cells among total blood leukocytes using flow cytometry (Attune, Life Technologies/Thermo Fisher Scientific). After engraftment confirmation (hCD45/total CD45 > 25%), mice were assigned to each treatment group by manual randomization based on their humanization rate, tumor volume and the cord-blood donor ID. The mice were then inoculated subcutaneously with 5 × 106 MDA-MB-231 cells resuspended in 50% Matrigel (Corning). When the tumors reached approximately 60 mm3 (day 19 after tumor cell inoculation), the animals were randomized into groups before initiating treatment. Mice received 30 mg kg−1 EOS301984 formulated in 2.5% DMSO, 10% solutol Hs-15 (Sigma-Aldrich) and 87.5% PBS twice a day intraperitoneally in a final volume of 200 µl, 10 mg kg−1 nivolumab in a final volume of 200 µl PBS intraperitoneally once every 5 days or a combination of both. All animals were monitored daily, checking for clinical signs of distress and recording body weight; tumors were measured three times a week using an electronic caliper. The tumor volume was calculated as volume = length × width2 × 0.5. Maximal tumor size was 2,000 mm3 and was not exceeded in the study.
Human specimensTumor samples were collected from individuals with colorectal cancer, renal cell carcinoma, NSCLC, endometrial cancer, melanoma or head and neck squamous cell carcinoma, all of whom voluntarily gave their informed consent according to the protocol approved by the local ethics commission at CNE KRD ‘Kyiv Regional Oncology Dispensary’ (Ukraine). Sample procurement and shipment were managed by BioPartners (USA; https://biopartners.science). In addition, NSCLC samples for the isolated TIL experiments were sourced by Fidelis Research (https://fidelis-research.com) according to the protocol approved by the National Commission for Bioethics of Medicines and Medical Supplies (Academia De Ştiinţe Medical, Romania). Cancer specimens were shipped at 4 °C in T-STORE medium (Life Science Production) and arrived within 48 h post surgery. Alternatively, samples were snap-frozen in liquid nitrogen for subsequent QMSI analysis. Patient characteristics are provided in Supplementary Table 1.
Venous blood from healthy volunteers, all of whom signed an informed consent approved by the Ethics Committee (FOR-UIC-BV-050-01-01 ICF_HBS_HD v.5.0), was obtained by Centre Hospitalier Universitaire Tivoli, La Louviere, Belgium. PBMCs were collected by density gradient centrifugation, using SepMate-50 tubes and Lymphoprep (STEMCELL Technologies) according to the manufacturer’s instructions. CD3+ T cells were isolated by immunomagnetic negative selection, using the EasySep Human T Cell Isolation Kit (STEMCELL Technologies) as per the manufacturer’s instructions. PBMCs and CD3+ T cells were stored in heat-inactivated FBS and 10% DMSO (Sigma-Aldrich) in liquid nitrogen. Alternatively, cryopreserved PBMCs derived from donors with a history of previous CMV infection and known HLA-A*02:01 expression were purchased from ImmunXperts (Gosselies, Belgium).
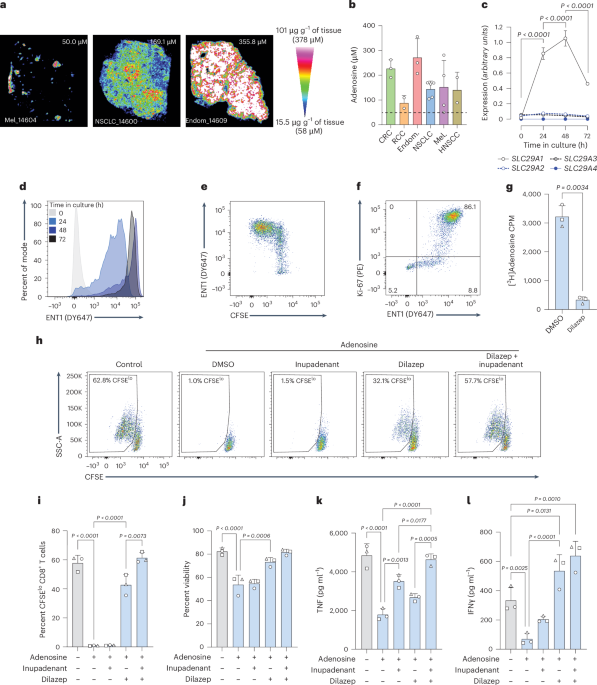
QMSI of adenosine in tumor tissueTumor tissues, stored at −80 °C, were placed inside the cryostat with the temperature maintained at −20 °C, and 10 µm tissue sections were obtained for each sample. Tissue cross-sections were placed on indium tin oxide (ITO) glass slides for MALDI imaging, and adjacent sections were collected on SuperFrost glass slides for H&E staining. The region of interest included tumor cells and possibly stroma and necrosis areas. Additional sections from a control sample were added for calibration curve purposes. Tissue sections on ITO slides were placed in a desiccator for 15 min. A solution of inhibitors was prepared to prevent the degradation of ADO in the tissue sections54 and was applied with a TM Sprayer (HTX imaging) on the sections immediately after the drying step in the desiccator and before any further treatment (calibrant deposit or spray of the MALDI matrix).
Two stable isotopic-labeled ADO (SIL-ADO) compounds were then considered in the next step for the QMSI for endogenous ADO, including a SIL-ADO calibrant as ADO-D2 and a SIL-ADO normalizer as ADO-13C5 (Toronto Research Chemical, cat. nos. A280409 and A280402, respectively), assuming a similar response of ionization between ADO and ADO-D2.
For quantification purposes, including the limit of detection, lower limit of quantification and upper limit of quantification, the different concentrations of ADO-D2 were deposited as follows: 1 µl of each ADO-D2 calibrant (including a zero calibrant) was spotted on the additional tissue sections and placed in a desiccator for 15 min before spraying the MALDI matrix on the entire slide. DAN (1,5-diaminonaphthalene; Merck, cat. no. 56451) was selected as the MALDI matrix for the detection of adenosine and was spiked with ADO-13C5 normalizer for spraying with the TM sprayer. The concentration of ADO-13C5 was adjusted to the expected signal of endogenous species in the tissue sections.
The MALDI-FTICR acquisition parameters were as follows: CASI mode; negative ionization; mass range, 272 ± 15 Da; laser frequency, 2,000 Hz; calibration mode, 2; spatial resolution, 130 µm for the regions of interest in the tumor sections with one region of interest selected per sample; 200 µm spatial resolution for the calibration range and 300 µm for the quality controls. Staining of adjacent or imaged sections (depending on their integrity after imaging) by H&E was prepared to overlay or compare the molecular images side by side with the histological structures.
MSI data were acquired and analyzed with FlexImaging, Data Analysis and the proprietary software MultImaging (v1.2.6.2). Pannoramic viewer (3D Histech) and ImageScope (Aperio) were used for histology.
Intensity scales representing the molecular signal were adjusted for each image to discriminate the noise from the molecular signal and to give the best visualization of the signal across the sections. A convolution step was performed on the original images using a normalized uniform kernel, which simply averages the values around a position. The kernel size was manually optimized for the analysis, minimizing the background noise.
Normalization of the data was performed with the stable ADO-13C5 based on an intensity ratio of the endogenous ADO (or ADO-D2 calibrant) and ADO-13C5 per pixel. A correlation between the calibration curve and the signal obtained on the tissues was then performed to determine the concentration of ADO per histological structure in µg g−1 of tissue or µM in the tissue sections.
Measurement of adenosine concentration in tumor interstitial fluidCell lines were subcutaneously inoculated into mice; when tumors reached ~300–400 mm3, they were removed and placed in nylon mesh with a pore size of 10 µm. The meshes containing the tumors were transferred to 1.5 ml tubes containing 10 μM pentostatin (Tocris) + 1 μM 5-iodotubericidin (Tocris). The samples were centrifuged at 450g for 10 min at 4 °C. The volume of fluid collected was measured and snap-frozen in dry ice and then stored at −80 °C. Total adenosine was quantified by liquid chromatography–mass spectrometry at Eurofins ADME Bioanalysis (France). In brief, calibration curves using a sample surrogate spiked with adenosine (Sigma-Aldrich) were used to set up a quantification method, with the limit of detection between 50 and 10,000 ng ml−1. Testing samples were prepared by using a QMA anion exchange cartridge to separate fractions with a potassium chloride salt gradient. After introducing QMA fractions into the cartridge, a centrifugation step at 100g was performed. Subsequent washes involved using 2 × 1 ml of 250 mM ammonium acetate followed by 2 × 1 ml of methanol. To elute the analytes, a solution of 20% formic acid in methanol was used, and the eluted material was then dried down and reconstituted in UP-water before injection into the liquid chromatography–tandem mass spectrometry system.
RNA extraction and real-time PCRCells were washed in cold PBS, resuspended in 350 µl of RLT+ buffer, mixed by pipetting up and down and vortexing for 30 s, and then the cell lysates were stored at −80 °C. RNA extraction was performed on all cell lysates at the same time, following the manufacturer’s protocol of the RNeasy plus micro kit from Qiagen (or mini kit for cell lysates with more than 1.5 × 106 cells). The concentration of RNA was measured using a NanoDrop One. Between 300 ng and 1 µg of RNA per sample was used for the reverse transcription step using the RevertAid kit from ThermoFisher Scientific according to the manufacturer’s instructions. A total of 10 ng of cDNA was loaded per well for the qPCR. TaqMan Fast Advanced Master Mix (10 µl per well) and the following Applied Biosystems TaqMan Gene Expression assays with FAM-MGB dye (1 µl per well; ThermoFisher Scientific) were used: SLC29A1 (assay ID, Hs01085706_m1), SLC29A2 (Hs01546959_g1), SLC29A3 (Hs00983219_m1), SLC29A4 (Hs00928283_m1), SLC28A1 (Hs00984391_m1), SLC28A2 (Hs01035846_m1), SLC28A3 (Hs00223220_m1), POLR2A (Hs00172187_m1) and SDHA (Hs00417200_m1). Nuclease-free water was added to reach a total volume of 20 µl per well. The plates were placed in a LightCycler 96 thermal cycler (Roche) for qPCR with the following steps: 95 °C for 20 s, 95 °C for 1 s and 60 °C for 20 s; steps two and three were performed 40× with a temperature ramp of 3.5 °C s−1. The assay was performed with a minimum of technical duplicates, and two housekeeping genes (POLR2A and SDHA) were included. These were selected based on stability of expression across samples as assessed with the geNorm application of qbase+ software (v.3.3). Normalization of expression values against the reference genes was performed on LightCycler 96 software (v.1.1.0.1320; Roche).
Western blotSnap-frozen cells were lysed in a modified RIPA buffer (Sigma-Aldrich) with 1× protease inhibitor cocktail (Thermo Scientific) by pipetting up and down, followed by 30 min incubation on ice. Samples were centrifuged for 10 min at 18,000g at 4 °C. The supernatant was transferred to a new microtube and stored at 4 °C. Protein concentration was quantified using a BCA protein assay kit (Pierce Thermo Scientific). Proteins were denatured at 95 °C for 5 min, separated electrophoretically using an Any kD Mini-Protean TGX gel (Bio-Rad) and then transferred onto 0.2 µM Nitrocellulose Filter Paper sandwiches (Bio-Rad). The membranes were blocked in 5% dry milk for 1 h and incubated with a specific primary antibody at 4 °C overnight. Blots were probed with rabbit anti-ENT1 (Abcam) and β-actin (Cell Signaling Technology) antibodies. After hybridization with horseradish-peroxidase-conjugated secondary antibody (Cell Signaling Technology), protein bands were visualized.
T cell activation culturesCryopreserved human CD3+ T cells were thawed and washed twice with RPMI 1640 medium, UltraGlutamine (Lonza) containing 10% FBS. CD3+ T cells were resuspended in PBS with 10% FBS at a concentration of 2 × 107 cells per ml. One volume of cells was combined with one volume of CFSE solution in PBS to give a final CFSE concentration of 1 µM. Alternatively, cells were incubated with eBioscience Cell Proliferation Dye eFluor 450 (final concentration, 4 µM). The cell suspension was mixed and incubated at 37 °C for 5 or 10 min, respectively, before cells were topped up with PBS with FBS (10%) to quench remaining excess labeling agent, then centrifuged at 450g for 5 min. T cells were resuspended in X-VIVO15 medium, and 5–8 × 104 cells were added to the wells of sterile round-bottom 96-well plates. Cells were activated by adding 50 μl of anti-CD3/CD28-coated microbeads (Dynabeads human T-activator CD3/CD28; Life Technologies), suspended in X-VIVO15 medium at a ratio of one microbead per two cells. Cells were cultured in the presence or absence of adenosine or ATP (Sigma-Aldrich) and additional reagents as indicated in the relevant figures. The final well volume was 200 µl in all cases, with DMSO concentration adjusted to 0.1% in all wells. Experiments were performed in technical duplicates. Cells were mixed by pipetting up and down and incubated for 3 days in a 37 °C humidified tissue culture incubator with 5% CO2.
In some experiments, GolgiPlug Protein Transport inhibitor (BD Biosciences) was added to T cell cultures (final dilution, 1:1,000) after 24 h of activation, and cells were cultured for a further 16 h before analysis of cytokine production by intracellular flow cytometry (see ‘Flow cytometry’ section).
CMV antigen recall assayCryopreserved human PBMCs were thawed and washed with RPMI 1640 medium containing 10% heat-inactivated FBS. The cells were suspended at 2 × 107 cells per ml in X-VIVO15 medium containing 5% (v/v) human serum and 1 mM sodium pyruvate. Then, 50 μl of cell suspension was added to the wells of sterile round-bottom 96-well plates. Cells were activated by adding CMV pp65 NLVPMVATV peptide (10 µg ml−1; day 0 of culture), IL-7 (days 0, 1 and 3 or 4 of culture) and IL-2 (day 3 or 4). A total of 300 μΜ of ATP was added to the cells in combination with 300 nΜ EOS301984 and/or 10 µg ml−1 anti-PD-1 or 10 µg ml−1 isotype anti-B-gal IgG4 in a 200 μl final volume. At day 1 and day 3 or 4, 100 µl of medium was removed and replaced with fresh medium including all compounds and cytokines at the original concentrations noted above. On day 6 or 7 of culture, cells were collected for flow cytometry analysis or rested before subsequent stimulation steps.
FluoroSpot analysisAfter 6 or 7 days of incubation, the cells from CMV cultures were washed twice with X-VIVO15 medium and left overnight at 37 °C in X-VIVO15 medium with 5% human serum and 1 mM sodium pyruvate. The following day, cells were processed for FluoroSpot analysis using the Human TNFa/IFNγ/Perforin/GranzymeB FluoroSpotFLEX kit (MABTECH) according to the manufacturer’s instructions. In brief, cells were counted and plated at either 5 or 10 × 104 cells per well in pre-coated FluoroSpot plates. NLVPMVATV peptide (10 µg ml−1) was added to the wells, with non-peptide-supplemented wells serving as controls, and plates were incubated overnight at 37 °C. The wells of the plate were then washed five times with PBS and incubated for 2 h at room temperature (20–25 °C) with the relevant detection antibodies. Plates were again washed five times with PBS and incubated for 1 h at room temperature with the relevant fluorophore conjugates. Following a further five washes with PBS, the fluorescence enhancer was added and the plate was left for 5–15 min at room temperature. The underdrain of the plate was removed and the plate left to dry in low light for a minimum of 24 h before being read on a FluoroSpot MABTECH IRIS reader (software v.1.1.45).
Cell killing assayThe lymphoblast cell line T2 was selected as a target for the killing assay based on HLA-A*02:01 expression, allowing presentation of the NLVPMVATV peptide. T2 cells were cultured in IMDM with 20% FBS, and on the day of the assay, the cells were washed in PBS and stained with cell proliferation dye eFluor 450 at final concentrations of 10 µM and 300 nM to create eFluor 450hi and eFluor 450lo populations, respectively. After a 10-min incubation at 37 °C in low light, 10 ml of cold IMDM with 20% FBS was added, and the cells were incubated for 5 min on ice. Cells were centrifuged and resuspended in 1 ml IMDM with 20% FBS. eFluor 450lo T2 cells were pulsed with NLVPMVATV peptide at a final concentration of 5 µg ml−1 and incubated for 2 h at 37 °C. Cells were washed twice and resuspended in X-VIVO15 containing 5% human serum and 1 mM sodium pyruvate at a final concentration of 1 or 2 × 105 cells per ml.
CMV-PBMC preparations were generated as outlined above and collected on day 6 or 7 of culture, washed and resuspended in X-VIVO15 with 5% human serum and 1 mM sodium pyruvate and left to rest overnight at 37 °C. On day 7 or 8, the pulsed and non-pulsed T2 cells and PBMC preparations were combined in different ratios. Based on the cell numbers available for the experiment, either 5 or 10 × 103 pulsed T2 cells were used and combined with an equal number of non-pulsed T2 cells and tenfold more PBMCs to give a ratio of 10:1 PBMCs to pulsed T2 cells. The mixture of cells was centrifuged for 20 s and incubated at 37 °C for 3 h. Cells were washed with PBS and stained with 400 nM ApoTracker for 15 min at room temperature in low light. Fixable viability eFluor780 was then added before a final incubation of 15 min at 4 °C. Cells were washed and resuspended in FACS buffer before acquisition on a BD LSRFortessa FACS machine.
Frequency of viable (viability dye− and ApoTracker−) T2 cells was determined, and the percent of viable pulsed T2 cells was divided by the percent of viable non-pulsed T2 cells, multiplied by 100 and then subtracted from 100 to generate the percent specific killing activity for each condition.
K d derivation for EOS301984 binding to human and mouse ENT1The Kd for EOS301984 binding to human and mouse ENT1 was determined in competitive binding experiments with [3H]HBMPR (Moravek, cat. no. MT-682) using membranes derived from JAR and MCA205-ENT2-KO cells, respectively.
Cells were amplified to mid-log phase in complete culture medium and then scraped from the culture vessels in ice-cold Ca2+-free and Mg2+-free PBS. Cells were centrifuged for 10 min at 5,000g at 4 °C, and the pellets were suspended in buffer A (15 mM Tris-HCl pH 7.5; 2 mM MgCl2; 0.3 mM EDTA; 1 mM EGTA) and homogenized in a glass–glass homogenizer. The crude membrane fraction was collected by two consecutive centrifugation steps at 35,000g and 4 °C for 30 min, separated by a washing step in buffer A. The final membrane pellet was suspended in buffer B (75 mM Tris-HCl pH 7.5, 12.5 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA, 250 mM sucrose) and flash-frozen in liquid nitrogen. Protein content was determined by the BCA method (Interchim, UP40840A).
The specific binding of [3H]NBMPR to ENT1 was confirmed in saturation binding experiments by incubating increasing concentrations of [3H]NBMPR with 10 µg of membrane preparation per well with the addition of unlabeled NBMPR (10 µM final assay concentration) or concentration-matched DMSO for 1 h at 4 °C in a 96-well plate. All components were diluted in assay buffer (Tris 50 mM, KCl 100 mM, MgCl2 0.1 mM, CaCl2 0.1 mM). The samples were then filtered over a GF/B filter plate (pre-incubated in 0.5% PEI (Sigma-Aldrich, cat. no. P3143) with a Filtermate Harvester (Perkin Elmer). After washing the filters five times with 0.5 ml of ice-cold assay buffer, 50 μl of Microscint 20 (Packard) was added to the filters, and the samples were incubated for 15 min on an orbital shaker and then counted with a TopCount for 1 min per well to derive CPM values. Bmax and Kd values for [3H]NBMPR specific binding (total binding − non-specific binding) were determined using the ‘One site – total and non-specific binding’ function in Graphpad Prism (v.10.0.2).
The kon for [3H]NBMPR was determined by incubating membrane extracts with a dose range of [3H]NBMPR for different times at 4 °C and then determining the CPM values as described above. The ‘One-phase association’ function in GraphPad Prism was used to derive the Kobs value for each concentration of [3H]NBMPR, which was then plotted against the matching concentration to derive the kon value (slope of the linear regression).
The koff for [3H]NBMPR was determined by pre-incubating 45 µl of membrane extracts with 45 µl [3H]NBMPR (10 nM final concentration) for 10 min until binding equilibrium was reached. A 200-fold excess of unlabeled NBMPR (10 µl) was then added to each well, and the CPM values were derived after various amounts of time as described above. The ‘Dissociation – one phase exponential decay’ function in GraphPad Prism was used to derive the koff value.
Competitive binding experiments were performed in which 50 µl of EOS301984 at various doses was combined with 25 µl of [3H]NBMPR (10 nM final concentration) and 25 µl of membrane extracts. Samples were incubated for 60 min at 4 °C, and CPM values were derived as described above. IC50 values for the effect of EOS301984 on the binding of [3H]NBMPR to ENT1 were determined with the ‘Log(inhibitor) vs response – variable slope (four parameters)’ function in GraphPad Prism.
Finally, 50 µl of EOS301984 at various concentrations above and below the IC50 value determined as above was incubated with 25 µl [3H]NBMPR (10 nM final concentration) and 25 µl membrane preparation at 4 °C for various periods of time. CPM values were generated as described above and used to generate kon and koff values for EOS301984 by using the ‘Kinetics of competitive binding’ function in GraphPad Prism55, in which the kon and koff values for [3H]NBMPR were constrained to those as derived above and Bmax as a shared value. Kd for EOS301984 was derived by dividing the koff by the kon values.
Polar metabolite measurement by liquid chromatography–tandem mass spectrometryT cell activation cultures were set up as outlined above (without CFSE staining). Following 24 h of culture, T cells were collected by combining three separate wells per condition into a single 1.5 ml microcentrifuge tube. Cells were pelleted by centrifugation at 4 °C for 5 min at 400g. Media was aspirated and cell pellets washed with chilled blood bank saline, followed by an additional centrifugation step at 4 °C for 5 min at 400g. Following aspiration of blood bank saline, cells were extracted in 80% MeOH/20% acetonitrile v/v with 250 nM 13C-labeled amino acids provided by the Whitehead Institute Metabolomics Core facility. Cells were extracted by vortex shaking in a 4 °C cold room, followed by centrifugation at 4 °C for 10 min at 16,000g to pellet insoluble material. The supernatant was transferred to a fresh microcentrifuge tube and dried down over gaseous nitrogen and stored at −80 °C before mass spectrometry analysis.
On the day of analysis, dried metabolites were resuspended in high-performance liquid chromatography-grade H2O. Metabolites were measured by liquid chromatography–mass spectrometry on a Q Exactive bench-top Orbitrap mass spectrometer equipped with an Ion Max source and a HESI II probe, which was coupled to a Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific). External mass calibration was performed using the standard calibration mixture every 7 days. From each sample, 4 μl was injected onto a SeQuant ZIC-pHILIC 150 × 2.1 mm analytical column equipped with a 2.1 × 20 mm guard column (both 5 μm particle size; EMD Millipore). Buffer A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide; buffer B was acetonitrile. The column oven and autosampler tray were held at 25 °C and 4 °C, respectively. The chromatographic gradient was run at a flow rate of 0.150 ml min−1 as follows: 0–20 min: linear gradient from 80–20% B; 20–20.5 min: linear gradient from 20–80% B; 20.5–28 min: hold at 80% B. The mass spectrometer was operated in full-scan, polarity-switching mode, with the spray voltage set to 3.0 kV, the heated capillary held at 275 °C and the HESI probe held at 350 °C. The sheath gas flow was set to 40 units, the auxiliary gas flow was set to 15 units and the sweep gas flow was set to 1 unit. MS data acquisition was performed in a range of m/z = 70–1,000, with the resolution set at 70,000, the AGC target at 1 × 106 and the maximum injection time at 20 ms. Relative metabolite quantification was performed in XCaliber QuanBrowser (v.2.2) (ThermoFisher Scientific) with 5 ppm mass tolerance, referencing an in-house library of chemical standards. Total ion counts for respective metabolites were exported into Microsoft Excel. To control for extraction efficiency, all samples within the same run were normalized to ion counts for 13C-labeled valine. To normalize metabolite levels to biomass, additional normalization was performed relative to unlabeled isoleucine. For each metabolite, the average of all three vehicle-controlled conditions was determined and used to determine log2(fold change) for the respective experimental conditions.
Tumor dissociation and cell cultureHuman tumor pieces were rinsed with cold PBS and cut into 1–2 mm fragments with a scalpel blade and transferred into GentleMACS C tubes. Dissociation of the tissue was completed with a Tumor Dissociation kit (Miltenyi Biotec) according to the manufacturer’s instructions. In brief, 5 ml of dissociation solution (200 µl of enzyme H, 20 µl of enzyme R and 25 µl of enzyme A, with the addition of RPMI to 5 ml) was added per gram of tumor. The tube was placed in the gentleMACS dissociator, and the 1st program was run. An incubation step of 30 min at 37 °C was applied with rotation (MACSmix tube rotator). After this incubation, a brief centrifugation (30g, 1 min) was performed, and the supernatant containing the dissociated cells was collected in a new tube containing FBS (30% of the total volume with supernatant) and placed at 4 °C. A second round of dissociation was performed on the remaining tissue fragments; after the addition of dissociation solution (same volume and content as the first round), the second gentleMACS program was run, and the tube was incubated for 30 min at 37 °C with rotation. Another brief centrifugation was performed, and the cells in the supernatant were pooled with the first collection and filtered through a 100 µm cell strainer, which was washed with 10 ml RPMI, 10% FBS and 2% penicillin–streptomycin. After centrifugation at 350g for 10 min, cells were resuspended in 20 ml RPMI, 10% FBS and 2% penicillin–streptomycin and counted with a haemocytometer. If cell agglomerates were visible, the cell suspension was filtered through a 100 µm cell strainer before counting. Dissociated tumor cells (DTCs) were either used directly for FACS analysis or washed into FBS with 10% DMSO and cryopreserved in liquid nitrogen until required.
Mouse tumors were digested as above with the following modifications: 100 µl of enzyme D, 50 µl of enzyme R and 12.5 µl of enzyme A were used in a final volume of 2.4 ml of RPMI per tumor. Processing was performed in the GentleMACS dissociator, followed by a 40 min digestion at 37 °C with shaking every 5 min. Samples were resuspended and passed through a 70 µm cell strainer over a 50 ml Falcon tube. The strainer was rinsed with 5 ml of RPMI, and the cells were centrifuged at 700g for 5 min and then resuspended in RPMI.
Cryopreserved human DTCs were thawed and washed twice with RPMI 1640 medium (Lonza) containing 10% heat-inactivated FBS. Cells were resuspended in PBS with 10% FBS at a concentration of 107 cells per ml. One volume of cells was combined with one volume of CFSE solution in PBS to give a final CFSE concentration of 1 µM. The cell suspension was mixed and incubated at 37 °C for 5 min before cells were topped up with PBS with FBS (10%) to quench remaining excess CFSE and then centrifuged at 450g for 5 min. The cells were resuspended at 4 × 106 cells per ml in X-VIVO15 medium containing 5% (v/v) human serum (Biowest), 1 mM Na-Pyr and 2% penicillin–streptomycin (Westburg/Lonza). Cell suspension (50 µl, 2 × 105 cells) was added to wells of sterile round-bottom 96-well plates. Cells were left unstimulated or were activated by adding IL-2 (50 U ml−1 final concentration, Proleukin-Novartis) with anti-CD3/CD28-coated microbeads (Dynabeads human T-activator CD3/CD28; Life Technologies) at a ratio of one microbead to five cells. ENT1 inhibitor EOS301984 (300 nM, iTeos Therapeutics; stock solution of 10 mM in DMSO) and anti-PD-1 (10 µg ml−1; OPDIVO (nivolumab), Bristol-Myers Squibb) were prepared and distributed to the relevant wells of the culture plate. A concentration-matched isotype control antibody was added to all wells not containing nivolumab, and DMSO concentrations were matched across all samples to 0.1%. Cells were cultured in the presence or absence of ATP (Sigma-Aldrich) at a final concentration of 500 μM. The final well volume was 200 µl in all cases. At a minimum, experiments were performed in technical duplicates. Cells were mixed by pipetting up and down and incubated for 3 days in a 37 °C humidified tissue culture incubator with 5% CO2. An additional dose of ATP at a final concentration of 500 μM was added 18 h after the start of culture.
For the summary analysis of T cell proliferation and cytokine production, the mean value of the readout recorded in the presence of ATP but with no other treatments was subtracted from all values. These were then expressed as a percentage of the mean value of control samples stimulated in the absence of ATP. This generated a scale of rescue from 0 to 100, in which 0 represents no difference from the ATP level and 100 represents a full rescue.
T cell populations isolated from cryopreserved DTCs (as outlined in the following section) were stimulated by culturing with anti-CD3/CD28 microbeads (one bead per two cells) at 2.5 × 104 cells per well in X-VIVO15 medium in the presence or absence of adenosine (100 µM) and EOS301984 (300 nM), as indicated in the relevant figure. Cells were processed for flow cytometry directly after 3 days of culture or resuspended in Cell Stimulation Cocktail (ThermoFisher Scientific) and incubated for 1 h at 37 °C. GolgiPlug Protein Transport inhibitor was added, followed by a further 4 h incubation before proceeding to flow cytometry.
For analysis of cytokine production by intracellular flow cytometry, mouse DTCs were stimulated at 1 × 106 cells per well in a 96 flat well plate in the presence of 0.4× Cell Stimulation Cocktail and 1× Protein Transport Inhibitor Cocktail (Fisher Scientific) in RPMI with 10% FBS and 50 µM β-mercaptoethanol for 3 h at 37 °C in a humidified incubator. Cells were centrifuged at 700g for 5 min at 4 °C before further processing for analysis by flow cytometry.
Flow cytometryCells were centrifuged at 450g for 5 min (used for all subsequent centrifugation steps), and the supernatant was discarded. For proliferation experiments (without ENT1 staining), cells were resuspended in PBS containing fixable viability dye and human Fc block (BD) and incubated for 15 min at 4 °C. For ENT1 staining, cells were resuspended in 50 µl NBMPR solution (20 µM, 0.02% DMSO; used as negative control for ENT1 expression) or DMSO (0.02% DMSO for ENT1 staining) as well as Fc block and then incubated 15 min at 4 °C; then, 50 µl of PBS containing SAHENTA-DY647 (0.2 µM for a final concentration of 0.1 µM) and fixable viability dye was added, followed by an incubation step of 30 min at 4 °C. Cells were washed in FACS buffer (PBS with 2 mM EDTA and 0.1% BSA) and resuspended in a solution of FACS buffer and relevant antibodies. After an additional 15 min of incubation at 4 °C, cells were centrifuged and resuspended in FACS buffer. In some experiments, cells were subsequently prepared for intracellular staining using the eBioscience Foxp3/Transcription Factor Staining Buffer Set (human cells); alternatively, IC fixation buffer and permeabilization buffers (mouse cells, eBioscience) were used according to the manufacturer’s instructions. Samples were analyzed on a BD LSRFortessa Cell Analyzer using BD FACSDiva software (v.9.0.1; Becton, Dickinson and Company) or a Cytek Aurora (SpectroFlo software, v.3.0.1). Analysis of FACS data, including uniform manifold approximation and projection, was performed using FlowJo (BD, v.10.8.1). Identification of discrete populations was performed manually; less well-defined populations were determined using fluorescence-minus-one controls or isotype control staining on matching DTC cell suspensions.
In some experiments, cryopreserved DTCs were thawed and washed with RPMI 1640 medium with 10% heat-inactivated FBS, washed in FACS buffer and resuspended at 8 × 107 cells per ml in PBS containing Fc receptor blocking solution. After 5 min at room temperature, cells were stained with fixable viability dye as outlined above, then washed in FACS buffer and resuspended in a solution of Brilliant Stain Buffer and relevant FACS antibodies. After a 20 min incubation at 4 °C, cells were washed twice in FACS buffer, resuspended at 1 × 107 cells per ml in FACS buffer and filtered through a 70 µm strainer. Sorting of PD-1− CD8+ T cells and PD-1+TIM-3+CD39+ CD8+ T cells was performed using a FACSAria II (BD FACSDiva v.9.0.1), and cell populations were collected in polystyrene tubes previously coated with FBS and containing X-VIVO15 medium with 1% penicillin–streptomycin. The purity of the sorted populations was assessed post-sort (Supplementary Fig. 7). Sorted cells were stored on ice until seeding.
A list of FACS antibodies and dilutions used in this study is provided in Supplementary Table 2.
Cytokine analysisCell culture supernatants were collected, and 5 µl of each was analyzed for TNF and IFNγ concentration using the TNFα (human) AlphaLISA Biotin-Free Detection Kit and IFN-γ (human) AlphaLISA Biotin-Free Detection Kit (Perkin Elmer), respectively, according to the manufacturer’s instructions, in 384-well OptiPlates (Perkin Elmer). Analysis was performed on a SpectraMax i3x (Molecular Devices) using SoftMax Pro 7 software (v.7.0.3; Molecular Devices). Alternatively, cytokines were analyzed with the Biolegend LegendPlex hCD8/NK kit according to the manufacturer’s instructions. The samples were run on a BD LSRFortessa and analyzed using Biolegend software (legendplex.qognit.com).
[3H]adenosine uptake assayHuman T cells were activated for 48 h with anti-CD3/CD28 microbeads in X-VIVO15 medium and then washed into ‘transport buffer’ (50 mM Tris-HCl pH 7.4, 120 mM NaCl, 3 mM K2HPO4, 10 mM glucose, 1 mM MgCl2, 1 mM CaCl2) before being dispensed in triplicate (2 × 105 per 25 μl) into the wells of a 96-well plate (Master Block, Greiner) containing 50 μl transport buffer or 50 μl dilazep diluted in transport buffer (1 μM final assay concentration). Cells were incubated for 15 min at room temperature before the reaction was initiated by the addition of 25 μl of a [3H]adenosine (Moravek Biochemicals)/adenosine mix (1:3) (1 μM final assay concentration). The reaction was incubated for 10 min at room temperature before rapid filtration through a GF/C SAN Unifilter plate pre-soaked in 0.5% PEI for 2 h at room temperature and washed five times with 500 μl of ice-cold transport buffer. Next, 50 μl of Microscint 20 (Packard) was added per filter, the plates were sealed, incubated for 15 min on an orbital shaker and then counted with a TopCount for 1 min per well. Alternatively, T cells were activated as described above for 48 h in the presence of a dose range of dilazep or EOS301984 and then centrifuged for 5 min at 450g before resuspension in transport buffer and used in adenosine uptake experiments as described above.
Nucleoside transporter inhibition assaysNucleoside transporter inhibition assays were performed using Madin-Darby canine kidney strain II (MDCKII) cells (German Cancer Research Center) stably expressing either human ENT1, ENT2, CNT1, CNT2 or CNT3; or HEK293 cells (Invitrogen) stably expressing human ENT4. Transfected cell lines were developed and characterized by SOLVO Biotechnology, a Charles River Company. Mock-transfected control and transfected cells were cultured in DMEM high glucose (4.5 g l−1) (Lonza) at 37 °C in an atmosphere with 5% CO2. Cells were plated onto standard 96-well tissue culture plates at a density of 1 × 105 cells per well. Uptake studies were performed 24 h after cell seeding.
Culture medium was removed and cells were washed twice with 100 μl of the respective assay buffer at pH 7.4 (Supplementary Table 3). Uptake experiments were conducted in duplicate at 25 ± 1 °C (CNT1, CNT2, CNT3 and ENT1 inhibition) or 37 ± 1 °C (ENT2 and ENT4 inhibition) in 50 μl of the respective assay buffer (pH 7.4). Cells were pre-incubated for 30 min with a concentration range of EOS301984 before addition of the appropriate probe substrate ([3H]uridine, [3H]adenosine (Moravek Biochemicals) or [3H]1-methyl-4-phenylpyridinium (MPP+; Perkin Elmer), and additional culture period as indicated in Supplementary Table 2. DMSO concentrations were equal in all wells and did not exceed 1.5% (v/v). After the experiment, cells were washed twice with 100 μl of appropriate cold assay buffer and lysed with 150 µl scintillation cocktail. Radio-labeled probe substrate transport was determined by measuring an aliquot (35 µl) from each well for liquid scintillation counting using a MicroBeta2 liquid scintillation counter (Perkin Elmer).
CPM values were corrected for non-specific transport by subtracting values obtained from control cells not expressing the relevant transporter and then transformed to a relative scale in which 100% is defined as transport in the absence of EOS301984. This value was subtracted from 100 to generate the percent inhibition of the relevant transporter.
Processing of published ChIP–seq, ATAC–seq and scRNA-seq dataNFAT ChIP–seq data were downloaded from NCBI Geo Set Omnibus (accession no. GSE116695 (ref. 13)) and visualized in the IGV browser56.
Human CD8-T ATAC–seq normalized count and raw fastq files were downloaded from NCBI Geo Set Omnibus (accession no. GSE89309 (ref. 34)). Raw fastq were aligned on human genome hg38 using bwa mem57 (v.0.7.18), and bigwig files were generated using bamCoverage from deepTools58 (v.3.5.4).
The Tumor Immune Cell Atlas was used to examine SLC29A1 and SLC29A2 expression across immune cells in multiple cancers52. The R package Seurat (v.5.1.0) was used for gene expression visualization59.
Oxygen consumption measurementsFollowing 24 h of culture, T cells were collected by combining three separate wells per condition into a single 50 ml conical tube. Cells were pelleted by centrifugation at 4 °C for 8 min at 400g. Media was aspirated and cell pellets were resuspended in unbuffered RPMI 1640 medium supplemented with 1 mM sodium pyruvate (Sigma-Aldrich). Then, 1.5–2.5 × 105 cells were plated into Seahorse XFe96/XF tissue culture plates (Agilent), previously coated with CellTak (Corning) according to the manufacturer’s instructions. Cells were allowed to recover in a 37 °C incubator without buffered CO2 before the Seahorse analysis.
For the Seahorse analysis, oxygen consumption measurements were determined (1) at baseline; (2) following oligomycin A injection (final concentration, 1.5 µM); (3) following FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) injection (final concentration, 2 µM); and (4) following antimycin A (all Sigma-Aldrich) and rotenone (Selleck Chemicals) injection (final concentration, 2 µM for both drugs). All values were normalized to total cell counts. Basal oxygen consumption rate was determined as the average of measurements at baseline minus the average of measurements following rotenone and antimycin A injection. Maximal oxygen consumption rate was determined as the average of measurements following FCCP injection minus the average of measurements following rotenone and antimycin A injection.
StatisticsStatistical analysis was performed with GraphPad Prism (v.10.0.2) using a combination of two-tailed paired t-tests, one-way or two-way ANOVA with correction for multiple comparisons and matching for samples as appropriate and indicated in the relevant figure legends. Dose–response curves were generated through the non-linear regression function ‘Log(inhibitor) vs response – variable slope (four parameters) and used to generate IC50 values (the concentration required for 50% maximal observed effect). Some additional statistics and data visualization were performed in RStudio (2023.03.1 Build 446).
Survival data were analyzed using the ‘Simple survival analysis (Kaplan–Meier)’ package in GraphPad Prism, with survival in mouse syngeneic tumor experiments defined as the time to reach the average tumor volume of the control group on the final day of measurement or 2,000 mm3, whichever was lower. For the xenograft experiments in humanized mice, survival was defined as the time to reach 500 mm3. Curves were compared using the log-rank (Mantel–Cox) test, and P values were corrected for multiple comparisons with the Bonferroni method.
No statistical methods were used to predetermine sample sizes. Sample size was chosen based on preliminary experiments or previously published results60. Data distribution was assumed to be normal, but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments. Randomization was performed for the xenograft experiment as described based on humanization rate and tumor volume, but not for syngeneic tumors in WT and ENT1-KO mice, as no treatment was applied. For in vitro experiments, all relevant treatments were applied to cells from each donor, so no randomization was required. No data were excluded from the analysis.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)