
Remember me




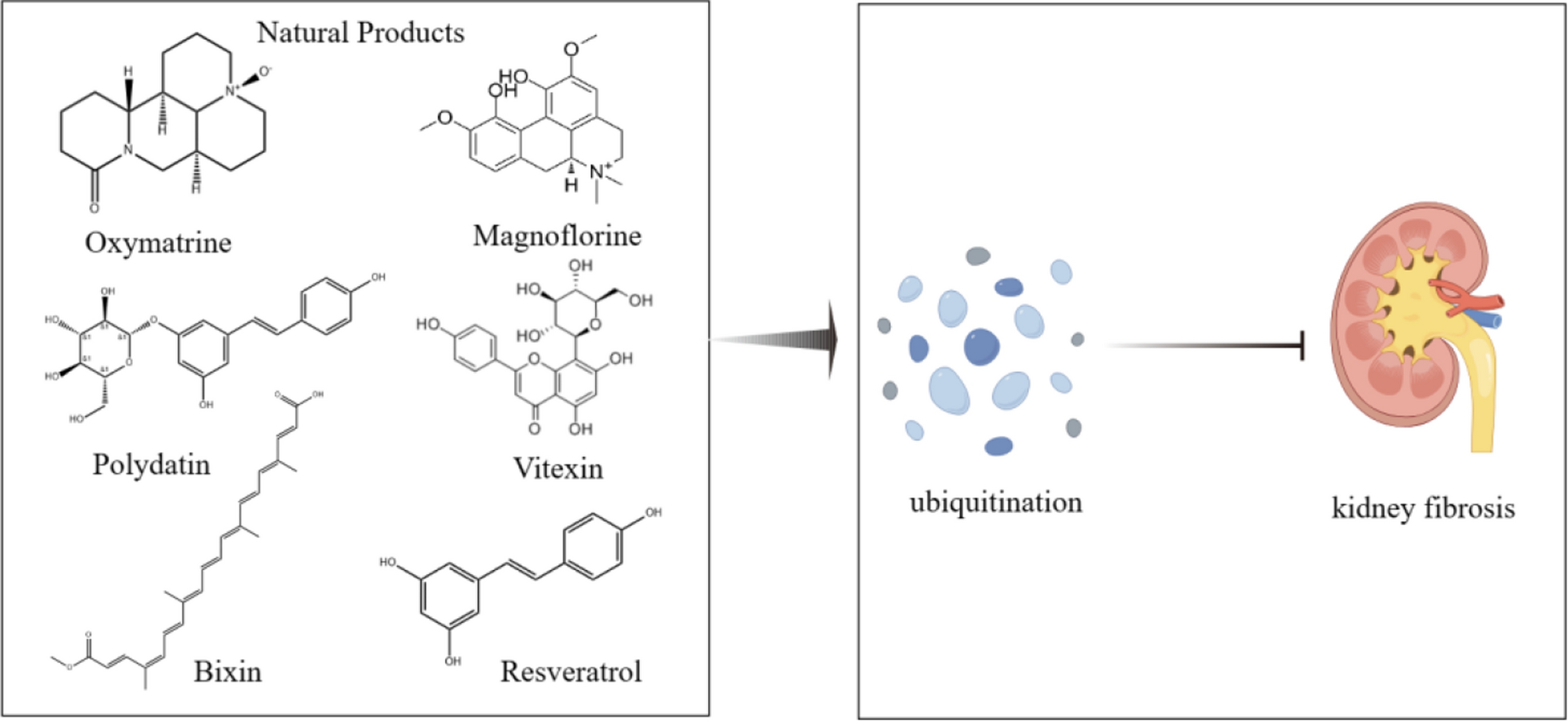
Renal fibrosis is the inevitable culmination of many progressive CKD, encompassing glomerulosclerosis and interstitial fibrosis, serving as a pivotal factor in the final stage of kidney disease [17]. Renal fibrosis is caused by a combination of inflammatory, growth factor, and lipid signaling pathways [18, 19]. According to research, the release of cytokines and chemokines following kidney damage promotes the activation of inflammatory cells and their infiltration into the kidneys. We must pay attention to them in order to stop renal fibrosis from getting worse. They include tumor necrosis factor-α (TNF-α), interleukin (IL), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and transforming growth factor-β (TGF-β) [20]. The infiltration of inflammatory cells and the persistent activation and proliferation of fibroblasts are key characteristics of renal fibrosis, which will ultimately result in the excessive buildup of extracellular matrix (ECM), the loss of parenchymal cells, and the eventual degradation of the kidney’s structural and functional integrity [21]. The process of epithelial-to-mesenchymal transition (EMT) in renal tubular epithelial cells (TECs), transforming into myofibroblast-like cells, is an important inducer of kidney fibrosis, highlighting its significance in the progression of the disease [22]. Research has identified numerous signaling pathways associated with renal fibrosis, among which TGF-β1/Smads, Wnt/β-catenin, JAK/STAT/SCOS, and Nrf2 are closely linked to ubiquitination (Figs. 1 and 2).
Fig. 1
The mechanisms and enzymes involved in ubiquitination and deubiquitination are illustrated in this figure, along with a number of natural compounds that enhance transcription factors and signaling pathways associated with kidney fibrosis through ubiquitination regulation. This figure was drawn with Figdraw (www.figdraw.com)
Fig. 2
This diagram generally visualizes the processes by which ubiquitination–deubiquitination modifications regulate the cell cycle, apoptosis, immune response, and DNA repair, including some important proteins and enzymes. This figure was drawn with Figdraw (www.figdraw.com)
TGF-β/Smads signaling pathwayTGF-β has a dual role in the kidneys, both with a profibrotic effect and possibly a protective effect [23]. Investigations have revealed that TGF-β plays a significant role in the regulation of ECM production and degradation, mesangial growth [24], and thickening of the glomerular basement membrane, primarily via the Smad or non-Smad pathways [25]. It mediates fibrosis by stimulating ECM production, inhibiting ECM degradation, and inducing the transformation of TECs into myofibroblasts [26]. Blocking TGF-β1 has been shown to prevent and ameliorate kidney fibrosis [27]. Research indicates that Types I and II TGF-β receptors are central to the most important for the transmission of TGF-β1/Smad signaling among types I, II, and III TGF-β receptors [28]. The Smad family comprises Smad1–8, with Smad2, Smad3, and Smad4 being closely linked to TGF-β1 signaling. Smad4 is critical for protecting the Smad complex, activating receptor-regulated Smads, and binding to target genes in the nucleus [29]. Evidence suggests a significant relationship between Smad proteins and the TGF-β1 signaling pathway, with Smad proteins interacting with other signaling pathways, thereby influencing the pathophysiology of kidney diseases [30, 31]. Fibrosis development is primarily facilitated by the TGF-β/Smads signaling pathway, which is triggered by ECM synthesis and accumulation, podocyte loss, mesangial expansion, tubular epithelial fibrosis, and the initiation of myofibroblasts [32]. Together with its receptors, TGF-β stimulates Smad2 and Smad3 [33], which in turn activate Smad4 [25, 34], and subsequently Smad7, α-smooth muscle actin (α-SMA), and so on [35, 36]. Smad7 interferes with TGF-β receptor binding by negatively regulating Smad2 and Smad3 activation, implicating Smad7 in fibrosis, cancer, and inflammation [37,38,39]. Ubiquitin ligases, Smurfs, are crucial in regulating the TGF-β/Smads signaling system, facilitated by the interaction between Smurf2 and Smad2, as well as by the selective targeting of Smad2 and transcriptional inhibitors in re with Smurf2 inducing ubiquitination and degradation of Smad1 [40]. Renal tubular epithelial cells are affected by renal fibrosis and EMT in a variety of ways, including modifications to cell phenotype, changes in cell function, and ECM modulation [41,42,43]. The significance of the TGF-β/Smads signaling pathway in the evolution of renal fibrosis is highlighted by recent findings that show overexpression of Smurf2 in the kidney causes ubiquitin-dependent degradation of Smad7, boosting TGF-β/Smad signaling and driving renal fibrosis [44].
Other proteins and processes, in addition to the ubiquitination of Smurfs, are crucial in controlling renal fibrosis brought on by the TGF-β pathway. By focusing on the ubiquitination and degradation of protein 53 (p53), tumor necrosis factor-α-induced protein 8 (TNFAIP8) enhances the survival and growth of fibroblasts, worsening renal fibrosis [45]. By breaking down Smad7, the E3 ubiquitin ligase Arkadia/ring finger protein 111 (RNF111) controls TGF-β signaling, which accelerates the development of fibrosis in a tubulointerstitial fibrosis model [46]. A lack of ubiquitin ligase neural precursor cell-expressed developmentally down-regulated 4-like (NEDD4-2) causes progressive kidney injury, which is characterized by renal tubular epithelial apoptosis, fibrosis, and other CKD symptoms [46].
Wnt/β-catenin signaling pathwayUnder damaged conditions, the complex and crucial Wnt/β-catenin signaling pathway is abnormally active [47]. The Wnt/β-catenin signaling system is involved in kidney development and fibrosis and regulates cell proliferation, survival, differentiation, and repair following kidney damage, among other aspects of renal physiology [48].Activation of the Wnt/β-catenin pathway regulates downstream EMT markers, increasing α-SMA expression while decreasing E-cadherin, thereby accelerating EMT progression and renal fibrosis [49]. Ubiquitination of β-catenin in its free state is a key step for degradation, controlled by the Wnt pathway [50]. When Wnt signals are absent, β-catenin is phosphorylated, but under stress or other stimuli, it remains unphosphorylated, accumulates, and activates target genes to regulate gene expression [51]. These studies validate that the upregulation of β-catenin ubiquitination can downregulate the occurrence of epithelial-to-mesenchymal transition (EMT), thereby intervening in the process of renal interstitial fibrosis. It has been shown that by raising β-catenin’s ubiquitination level, the nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) can affect the expression of the Wnt/β-catenin signaling pathway [52]. Renal fibrosis can be slowed down by suppressing the traditional Wnt/β-catenin pathway, as this system increases the extracellular matrix (ECM) and causes extracellular matrix activation [52]. In the model of chronic kidney disease (CKD) brought on by ischemia–reperfusion injury (IRI), doxorubicin, or unilateral ureteral obstruction (UUO), inhibiting Wnt/β-catenin signaling can improve renal function, decrease collagen deposition and renal fibrosis, and inhibit the expression of genes linked to fibrosis [48].
The Notch and Hedgehog pathways are among the signaling pathways with which the Wnt/β-catenin pathway interacts. By creating a complex with β-catenin, Notch facilitates the breakdown of β-catenin, which in turn prevents the differentiation of epithelial cells and accelerates the development of renal fibrosis [53]. The Hedgehog and Wnt/β-catenin signaling pathways overlapped and worked in concert, and the Hedgehog pathway influenced the Wnt/β-catenin route, which in turn influenced the development of renal fibrosis [53].Negative regulators like Adenomatous Polyposis Coli (APC), Glycogen Synthase Kinase-3 (GSK-3), and Axin regulate the Wnt/β-catenin signaling pathway [54]. When these negative regulators are mutated or deleted, β-catenin builds up in the cytoplasm and moves to the nucleus, where it combines with other transcription factors to activate downstream target genes, advance the cell cycle, and trigger EMT [54]. Furthermore, other EMT-related transcription factors, including Snail, Twist, and ZEB1/2, interact with the Wnt/β-catenin signaling pathway. These transcription factors are triggered by a variety of signaling pathways, including TGF-β, Wnt/β-catenin, Hedgehog, and Notch [55].Certain E3 ubiquitin ligases, such as beta-transducin repeat-containing protein (β-TrCP), JNK-associated leucine zipper protein downstream of c-Jun (Jade1), Casitas B-lineage lymphoma (c-Cbl), tripartite motif-containing protein 33 (Trim33), siah E3 ubiquitin protein ligase 1 (SIAH1), E3 ubiquitin ligase identified by differential display (EDD) [56], and tribbles pseudokinase 2 (TRIB2), mediate the ubiquitination of β-catenin [57]. These ligases are important regulators of Wnt signaling pathways and impact a number of biological processes, including cell proliferation, differentiation, and carcinogenesis, by influencing the stability and function of β-catenin (Table 1).
Table 1 Natural products improve renal fibrosis by modulating ubiquitination modificationsJAK/STAT/SOCS signaling pathwayThe JAK/STAT signaling pathway is intricately linked to the regulation of inflammation, cell proliferation, and fibrosis, with SOCS proteins serving as negative regulators of the JAK/STAT pathway [58, 59]. The transcription factors that make up the STAT family are STAT1 through STAT6, with STAT3 having a more significant function in the JAK/STAT pathway [60]. Research findings indicate that STAT6, a transcription factor that is degraded by the liposome and ubiquitin proteasome pathways, is a promoter region where TGF-β1 binds to a gene [61]. By controlling the ubiquitination and degradation of STAT6, the ubiquitin–proteasome system influences the activity of STAT6 during the fibrosis process [62]. It then takes part in the regulation of immune response and cell metabolism, both of which have a significant influence on the onset and progression of fibrosis [62].
Research has demonstrated that the expression of JAK1, JAK2, and STAT3 was up-regulated and the expression of Stat4 was down-regulated in the diabetic nephropathy model, and the activation of SOCS1/3/7 can reduce urinary albumin levels in mice and ameliorate renal fibrosis by means of the JAK/STAT/SOCS signaling pathway [58]. A major factor in diabetic nephropathy (DKD) is the JAK/STAT signaling system, which is activated by hyperglycemia and the vasoactive peptide ANG II. This results in increased mesangial cell proliferation and growth, which in turn promotes renal fibrosis [63]. Furthermore, the pathophysiology of DKD is intimately linked to podocyte autophagy damage, which is primarily mediated by the JAK/STAT signaling system [64]. Activation of this pathway can enhance apoptosis and block podocyte autophagy, exacerbating podocyte injury and the development of DKD [64]. In summary, the JAK/STAT signaling system is critical for the kidneys, particularly in diabetic nephropathy, where it activates genes that are pro-inflammatory and profibrotic, hence promoting renal fibrosis [65]. The JAK/STAT pathway, in particular the STAT3 branch linked to renal fibrosis, can be activated by diabetes mellitus [65]. Renal fibrosis is characterized by the buildup of extracellular matrix proteins and the conversion of kidney cells into myofibroblasts, which can be facilitated by STAT3 activation, which can also result in the transcription of profibrotic genes [65].
Nrf2 signaling pathwayThe stimulation of antioxidative genes and the activation of various antioxidants depend on the transcription factor Nrf2, which helps to maintain redox equilibrium and eliminate the buildup of ROS [66]. Redox maintenance enzymes such as Kelch-like ECH-associated protein 1 (Keap1) and heme oxygenase-1(HO-1) can regulate the activation of NRF2 [67]. Controlling cell metabolism and inflammation is one of the Nrf2-Keap1 signaling pathway’s primary roles. Based on experimental findings, Keap1 may be able to control Nrf2’s ubiquitination, which will cause the 26S proteasome to recognize and degrade it, keeping it in the cytoplasm and inhibiting its nuclear translocation [68]. Conversely, in answer to electrophilic stimuli, the Nrf2-Keap1 complex is broken, which stops Nrf2 ubiquitination and permits Nrf2 to go to the nucleus, where it increases [69, 70]. This is a significant difference in both preventing and treating renal fibrosis.
When it comes to renal fibrosis, the Nrf2 signaling pathway is significant. The Nrf2 signaling pathway inhibits the progression of renal fibrosis by reducing oxidative stress and inflammatory responses through the activation of downstream antioxidant and anti-inflammatory gene expression. In particular, by controlling the phosphatidylinositol 3-kinase/protein Kinase B (PI3K/Akt) signaling pathway, the Nrf2 signaling pathway can block the epithelial–mesenchymal transition, halt the progression of kidney disease, and decrease renal tubular epithelial cell death and interstitial fibrosis in a diabetic nephropathy model [71]. Furthermore, elevated expression of catalytic and antioxidant enzymes like HO-1 in renal tubular epithelial cells, which can scavenge reactive oxygen species and shield the kidney from oxidative injury, is linked to Nrf2 signaling pathway activation [72]. These studies offer a solid foundation for the creation of kidney disease therapy plans that specifically target the Nrf2 signaling pathway, as well as significant insights into the mechanism of action of this pathway in kidney disease.
Other signaling pathwaysHowever, recent studies highlight the crucial role of ubiquitination modifications in the progression of renal fibrosis, and there are currently some studies exploring the role and mechanism of ubiquitination modification of proteins in renal fibrosis. Under certain pathological conditions, beyond the TGF-β1/Smads, Wnt/β-catenin, JAK/STAT/SOCS, and Nrf2 pathways, other pathways such as Connexin32-NADPH oxidase 4 (Cx32-Nox4) and Insulin-like growth factor 1 receptor (IGF-1R) are also connected to ubiquitination-regulated pathophysiological changes in renal fibrosis. The pathophysiological alterations of renal fibrosis are significantly influenced by IGF-1R and Cx32-Nox4. According to preclinical research, Polygonum cuspidatum reduces the expression of FN and ICAM-1, the primary factors that worsen diabetic renal fibrosis, by controlling the Cx32-Nox4 signaling pathway, lowering Nox4 protein levels, and reducing reactive oxygen species production by encouraging K48-linked polyubiquitination of Nox4 [73]. Icariin has also been shown to lower renal oxidative stress levels and the production of fibrotic proteins, as well as improve hypertensive renal fibrosis and damage via the Cx32-Nox4 signaling pathway [74]. By controlling the expression of IGF-1, which can promote kidney cell proliferation and protect renal tubular epithelial cells in rats with kidney injury, the Heidihuang pill inhibits renal fibrosis in rats with chronic renal failure in terms of IGF-1R [75]. These studies demonstrate the potential utility of Cx32-Nox4 and IGF-1R in the therapy of renal fibrosis by presenting preclinical and clinical evidence for their regulatory roles in the disease.
Comments (0)