
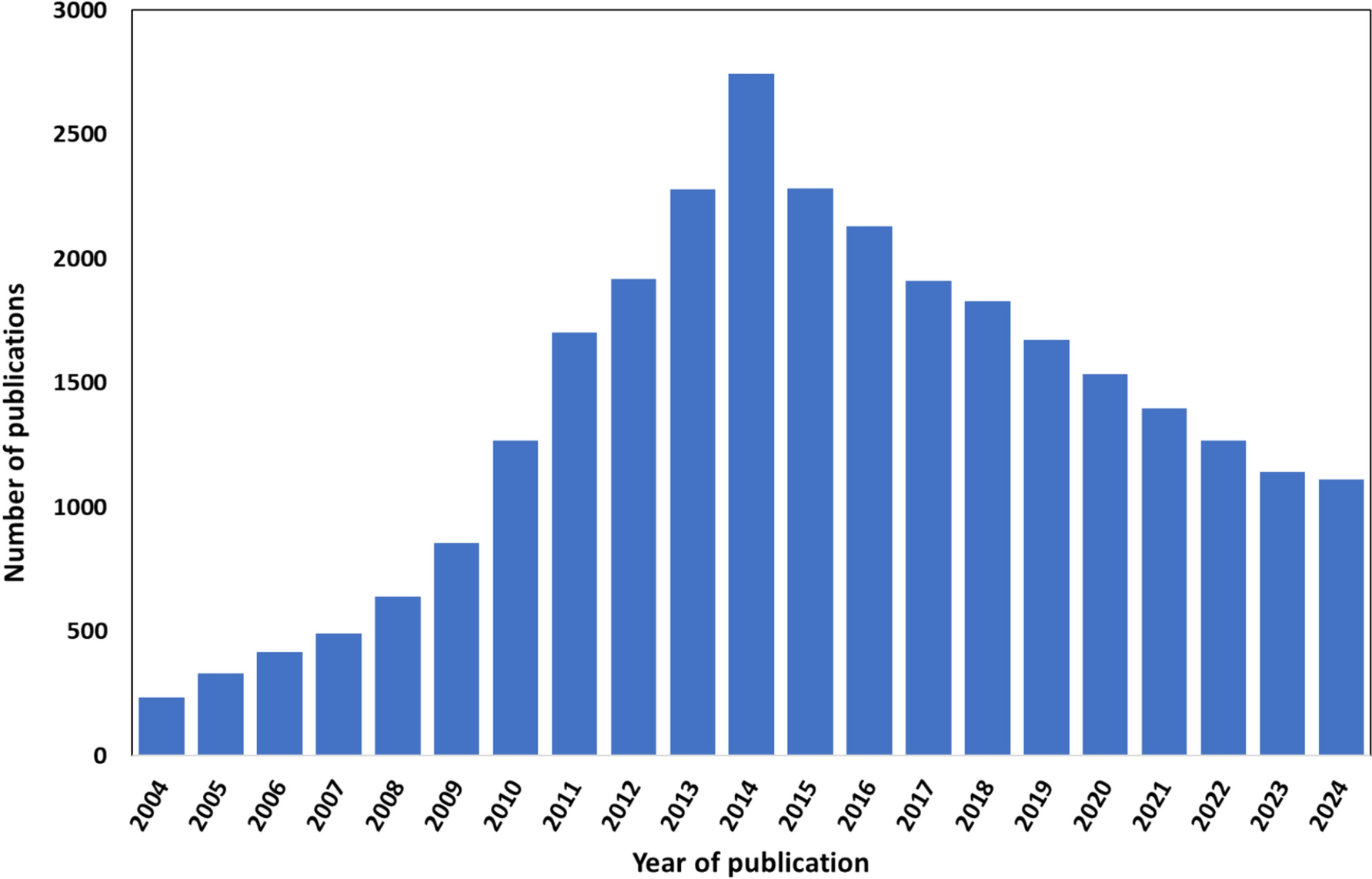
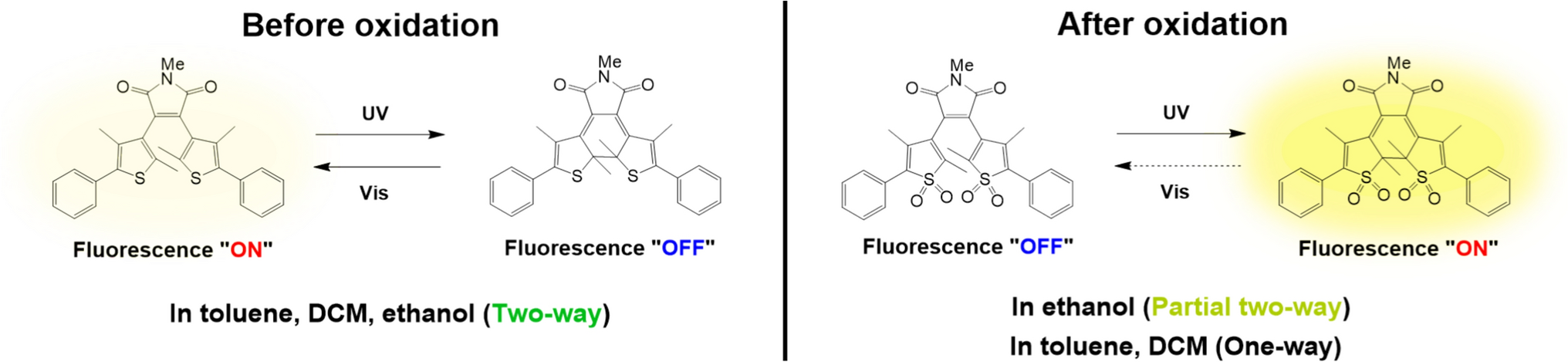
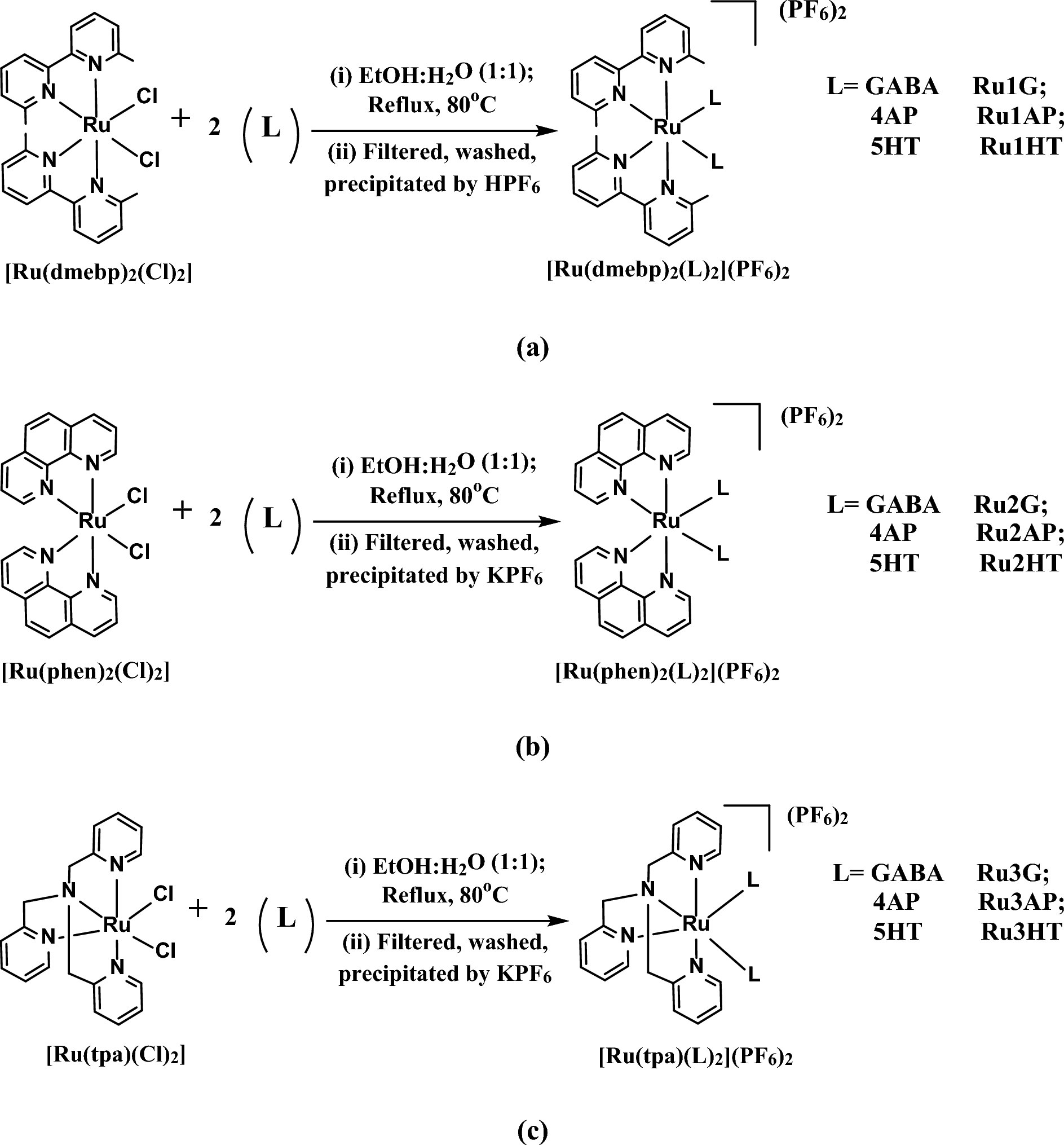
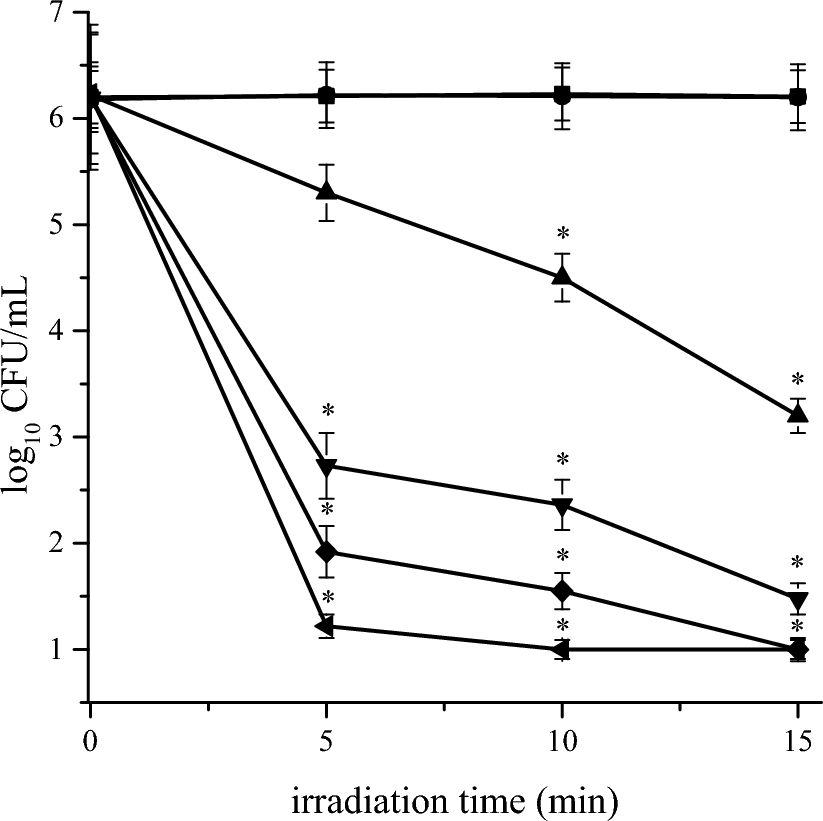
Remember me



The ε-adducts reactivity toward 1O2 was first considered using rose Bengal (RB2−) as a typical Type II photosensitizer [18, 19]. Steady-state photolysis was run for aerated water solutions of the ε-adducts (20 µM) and RB2− (10 µM) using white light (emission between 350 and 700 nm) to ensure the excitation of RB2− at its 550 nm absorption band without direct excitation of the ε-adducts (Fig. S1). The kinetics of the lesion consumption and photoproducts formation were followed by HPLC. Control experiments were performed by irradiating the lesion alone; expectedly, no changes were observed by HPLC confirming their photostability under our experimental conditions (Fig. S2). Conversely, steady-state photolysis of the ε-adducts in the presence of RB2− showed that the initial peak decreases as a function of irradiation time (Figs. 2 and 3, black lines).
Fig. 2
a HPLC chromatograms registered at 260 nm for an aerated solution of 20 µM εdG in the presence of 10 µM RB upon irradiation at different times in H2O. B Variation of the concentration of damaged (εdG, circle) and repaired nucleoside (dG, square) as a function of the irradiation time in H2O (black) and D2O (red)
Fig. 3
a HPLC chromatograms registered at 260 nm for an aerated solution of 20 µM εdA in the presence of 10 µM RB upon irradiation at different times in H2O. B Variation of the concentration of damaged (εdA, circle) and repaired nucleoside (dA, square) as a function of the irradiation time in H2O (black) and D2O (red)
The guanine-derived adduct, εdG (eluting at 15 min), exhibited a reactivity similar to that previously reported in the literature [17] as its consumption gave rise to the formation of different products with retention times (tR) of 5.2–5.4, 6.2, and 10.7 min (Fig. 2). Compound eluting at 6.2 min was assigned to the original nucleobase, dG, by comparison with an authentic sample. Compounds at 5.2–5.4 and 10.7 min were identified by HPLC–MS (Fig. S3) on the basis of Martinez et al. studies [17]. The fast eluting compound with m/z of 326 corresponds to intermediate III, whereas at tR = 10.7 min the registered m/z of 296 agrees with the generation of intermediate VIa and/or VIb (Scheme 1).
Interestingly, εdG is not the only ε-adduct that reacted under our experimental conditions. As shown in Fig. 3, εdA (eluting at 11.2 min) was also consumed, but led to the formation of one single product eluting at tR of ca. 8.5 min and assigned to dA by comparison with an authentic sample and HPLC–MS analysis. In that case, no intermediates of the reaction were detected.
To confirm the role of 1O2 in the degradation of the ε-adducts and regeneration of the original DNA bases, irradiations were run in D2O, where 1O2 lifetime is much longer than in H2O (ca. 60 vs 4 µs) [22]. Accordingly, the kinetics were faster, reaching an almost complete consumption after 20 and 60 min for εdG and εdA, respectively (Figs. 2B and 3B, red lines).
Thus, RB2− photosensitization of both ε-adducts under aerobic conditions leads back to the original nucleosides and provides a photorepair process of these lesions. Recovery of the nucleosides appears to occur in high yields for εdA in D2O (Fig. 3B). By contrast, dG formation yield reaches less than 10% just after 15 min of irradiation in D2O. This low yield can be explained not only by the presence of intermediates that, as shown by Martinez et al., slowly evolve to dG, but also by the particular reactivity of dG with 1O2, as it is the only DNA nucleobase able to react with this reactive oxygen species to yield secondary products [23].
2.2 Photophysical study of RB2− in the presence of ε-adductsNext, the interaction of the ε-adducts with 1O2 was studied using time-resolved phosphorescence. Experiments were run recording 1O2(1Δg) characteristic emission at 1270 nm after excitation at 532 nm of D2O solutions of RB2− at a concentration of 5 µM (A532 = 0.18). The initial lifetime τ0 of ca. 66 µs determined for 1O2 emission was shortened after the addition of increasing amounts of the etheno derivatives (Fig. 4). The 1O2 quenching rate constants (kq(1O2)-ε in M−1s−1) were determined from the Stern–Volmer plots applying the following equation:
$$1/\tau = 1/\tau_ + k_ \left( ^}}_} } \right) - \varepsilon \times \left[ }} \right],$$
Fig. 4
Time-resolved 1O2 phosphorescence signal (λexc = 532 nm, λem = 1270 nm) and their corresponding Stern–Volmer plot (insets) obtained from aerated D2O solutions of 5 µM RB in the presence of increasing amounts of ε-adducts (A) from 0 to 2 mM for εdA and (B) from 0 to 0.2 mM in case of εdG
where τ0 and τ are the 1O2 lifetime (in s) in the absence and in the presence of a concentration [ε-adduct] of the quencher (in M). Similar values were obtained for the two adducts with kq(1O2)-ε in the order of 106 M−1s−1 (Table 1).
Table 1 Quenching rate constants of 1O2, generated after RB2− excitation, by ε-adducts (kq(1O2)-ε), bimolecular quenching rate constants of 3RB2−* by ε-adducts determined from phosphorescence, (kq(3RB2−*)P-ε) and from laser flash photolysis (kq(3RB2−*)LFP-ε) experiments, and bimolecular quenching rate constants of 3CBP* by ε-adducts determined from laser flash photolysis (kq(3CBP*)LFP-ε) experimentsMoreover, a decrease of the top emission intensity was observed for all the additions, which was especially pronounced for εdA (Fig. 4A) where the final adduct concentration was higher (εdG is not soluble at concentration higher than 0.4 mM). This decrease in 1O2 signal intensity could be the result of a lower formation yield of the 1O2 precursor, i.e., RB2− triplet excited state (3RB2−*), as the concentration of the quencher increases. These changes in 3RB2−* population can be, indeed, due to several processes.
First, the screening effect resulting from absorption of the etheno derivatives can be discarded due to their lack of absorption at the excitation wavelength (λexc = 532 nm). Another option is an interaction of the ε-adducts with the singlet excited state of the photosensitizer (1RB2−*), which would decrease the excited molecules available for intersystem crossing and population of 3RB2−*, resulting in less 1O2 production. To investigate this possibility, steady-state fluorescence experiments were carried out under the same conditions as those of 1O2 detection, but no significant decrease of 1RB2−* emission intensity was observed (Fig. S4). Therefore, on the basis of the steady-state fluorescence results, the decrease in the top emission intensity observed in Fig. 4 is not due to a quenching of 1RB2−*. Another explanation could rely on the photodegradation of the RB2− during the measurements (each trace is obtained after an accumulation time of 3 min). However, no significant changes were observed when comparing the UV–Vis absorption spectra of the solutions before and after the 1O2 measurement.
A direct interaction between the triplet excited state of RB2− and the ε-adducts represents another possibility. Indeed, upon excitation, formation of the triplet manifold is the main route of 1RB2−* deactivation with an efficient intersystem crossing, \(\phi\)ISC of ca. 0.8, and thus the reactivity of this state directly with the lesions can play an important role in the photochemical processes [19, 24, 25]. In this context, phosphorescence experiments were performed using RB2− solutions (5 µM) prepared in deuterated water and flushed with N2 just before the measurements. The phosphorescence emission was registered at room temperature after excitation at 532 nm and showed a maximum at 730 nm with a lifetime τ0 of ca. 210 µs. Decays were recorded in the presence of increasing amounts of the ε-adducts (Fig. 5), and the bimolecular quenching rate constants kq(3RB2−*)P-ε, summarized in Table 1, were obtained from the Stern–Volmer plots (Fig. 5, insets). These phosphorescence experiments showed that 3RB2−* is quenched by εdA and εdG, with a constant two orders of magnitude higher for the latter. Interestingly, for εdG, the quenching rate constant of 3RB2−* is also two orders of magnitude higher than that of 1O2.
Fig. 5
Time-resolved 3RB2−* phosphorescence signals monitored at 690 nm in N2 obtained from 5 µM RB2− in the presence of increasing amounts of ε- adducts: A εdA and B εdG (from 0 to 2 mM and 0 to 0.2 mM in the case of εdG). Insets: corresponding Stern–Volmer plot
On these bases, laser flash photolysis experiments were carried out to get more insight into the quenching mechanism and to investigate the presence of other intermediates such as radical species in the ε-adducts photosensitization process. A previous work revealed that 532 nm laser excitation of RB2− leads not only to the formation of its triplet excited state, but also to oxidative and reductive processes leading to the corresponding ion radicals (Fig. S4) [18]. Thus, experiments were performed by means of a nanosecond pulsed laser (Nd:YAG) using 532 nm as the excitation wavelength. The transient absorption spectra of a degassed solution of RB2− in D2O at different times after the laser pulse are in agreement with those described in the literature for RB2− [18], and provide evidence for the formation of three transient species (Fig. S4). The first one, exhibiting three maxima at \(_}^}\) 380, 465 and 590 nm, is attributed to the triplet–triplet transition 3RB2−* that starts to decay just after the pulse has a lifetime of τ(3RB2−*) = 82 µs. The other two transients are longer-lived than 3RB2−* and appear at \(_}^}\) 420 and 460 nm with a relative delay after the pulse. On the basis of literature data, they were assigned to the oxidized (RB•−) and reduced (RB•3−) forms.
The typical RB2− signals were obtained in the presence of both ε-adducts (Fig. 6), and quenching experiments were run by adding increasing amounts of the different etheno derivatives. For the three lesions, the signal at ca. 600 nm assigned to the triplet–triplet transition was monitored, and bimolecular quenching rate constants of the same order of magnitude as those obtained during the luminescence studies were determined. The obtained values are kq(3RB2−*)LFP-ε of ca. 3.8 × 106 and 8.5 × 107 M−1s−1 for εdA and εdG, respectively (Table 1). Interesting outcomes can be drawn from the ion radical signals. As shown in Fig. 6, when εdA and εdG were added to the RB2− solution, the signals at 380, 465 and 590 nm decay, whereas the transient absorption corresponding to the photosensitizer reduced form RB•3− at 420 nm remains stable. For further assignment, formation of RB•3− was forced by adding 1,4-diazabicyclo[2.2.2]octane (DABCO) to the system. Figure S6 shows the transient absorption spectra of RB2− in the presence of 5 mM DABCO, where the RB•3− signal at 420 nm is clearly observed. Therefore, detection of this species when εdG and εdA are present in the solution, together with 3RB2−* quenching by these ε-adducts, points toward a photoredox process where 3RB2−* oxidizes the lesions to yield the photosensitizer reduced form, RB•3−, and the cation radical of the lesions. This latter was not detected in our experiment; however, it could be masked by the band derived from RB2−, have a low molar absorption coefficient or absorb in a region different from the analysis window.
Fig. 6
Transient absorption spectra of a deaerated solution of RB2− in deuterated water after 532 nm excitation in the presence of 2 mM of εdA (A) and 0.2 mM of εdG (B). Insets: Stern–Volmer plot obtained from decays at 600 nm
2.3 Photosensitization of ε-adducts by CBPIn view to study a potential electron transfer process leading to ε-adducts oxidation, 4-carboxybenzophenone (CBP) was considered as a Type I photosensitizer. First of all, steady-state photolysis experiments were done to investigate the occurrence of the repair process. The ε-adducts were irradiated with UVA light in the presence and absence of CBP in deaerated water. Experiments were performed in the absence of oxygen to avoid formation of 1O2 and focus attention on a potential electron transfer process. No degradation of the etheno derivatives occurred during the control irradiation in the absence of CBP. By contrast, in the presence of CBP, photodegradation took place for the two etheno derivatives (Fig. 7). For εdG, the same pattern as that in Fig. 2A was observed in the chromatograms, where the repaired nucleoside was observed together with other peaks at a tR of ca. 5.2–5.4 and 10.8 min (Fig. 7A). By contrast for εdA, in addition to dA, other photoproducts were formed at short elution times, between 3 and 5 min (Fig. 7B). Due to the low concentration of these compounds, no clear information on their nature was obtained from HPLC–MS analysis.
Fig. 7
HPLC chromatograms registered at 260 nm for an anaerobic H2O solution (1.5% acetonitrile) of A εdA or B εdG (100 µM) in the presence of CBP (50 µM) upon UVA irradiation at different times. Inset: variation of the concentration of ε-adduct (black square) and repaired nucleoside (red circle) with irradiation time
These results point toward the possibility of a photoinduced anaerobic oxidation process for the purine derivatives, which regenerates the original nucleobase from the ε-adducts without intervention of oxygen. In that case, and by contrast with Type II photosensitization, once formed both dA and dG are sensitive to electron transfer, which can lead to the formation of oxidatively generated photoproducts.
2.4 Photophysical study of CBP in the presence of ε-adductsLaser flash photolysis experiments were carried out at 355 nm to investigate the nature of the observed reactivity. The transient absorption spectra of a N2 flushed CBP aqueous solution showed two signals at 340 and 560 nm assigned to the CBP triplet excited state (3CBP*) bands by comparison with reported data (Fig. S7) [20]. In the presence of purine-derived ε-adducts, the 3CBP* disappeared at short delays after the laser pulse and gave rise to a longer-lived and red-shifted signal with maximum at ca. 580 nm (Fig. 8). This behavior was more pronounced in the case of εdG than for εdA. The new transient was attributed to the ketyl radical of CBP obtained from photoreduction of the 3CBP* and oxidation of the lesions. In this process, the formed anion radical CBP•− is subsequently protonated (Scheme S2) [26] yielding CBPH•. This transient radical was further assigned by comparison with the signals obtained for CBP aqueous solution containing 20% of ethanol (inset Fig. S7). In this case, the typical H-abstraction from the solvent led to efficient formation of CBP ketyl radical centered at 580 nm.
Fig. 8
Transient absorption spectra of CBP in the presence of ε-adduct (2 mM for εdA and 0.3 mM for εdG) in MeCN:H2O (1:1, v:v) at different times after the 355 nm laser pulse. A εdA, B εdG
Concerning the kinetics, the 3CBP* lifetime was shortened in the presence of the two etheno derivatives. The bimolecular quenching rate constants kq(3CBP*)-ε obtained from the Stern–Volmer plots were of ca. 3.6 × 109 M−1s−1 for εdG and 2.2 × 109 M−1s−1 for εdA (Table 1). Interestingly, the kq(3CBP*)-ε was higher in the case of εdG, which suggests, as reported for the canonical base, a lower oxidation potential of the guanosine derivative with respect to εdA. Altogether, these data are consistent with quenching occurring via electron transfer from the ε-adduct to CBP triplet manifold, and it is further supported by the clear detection of CBPH• in Fig. 8.
Comments (0)