
Remember me

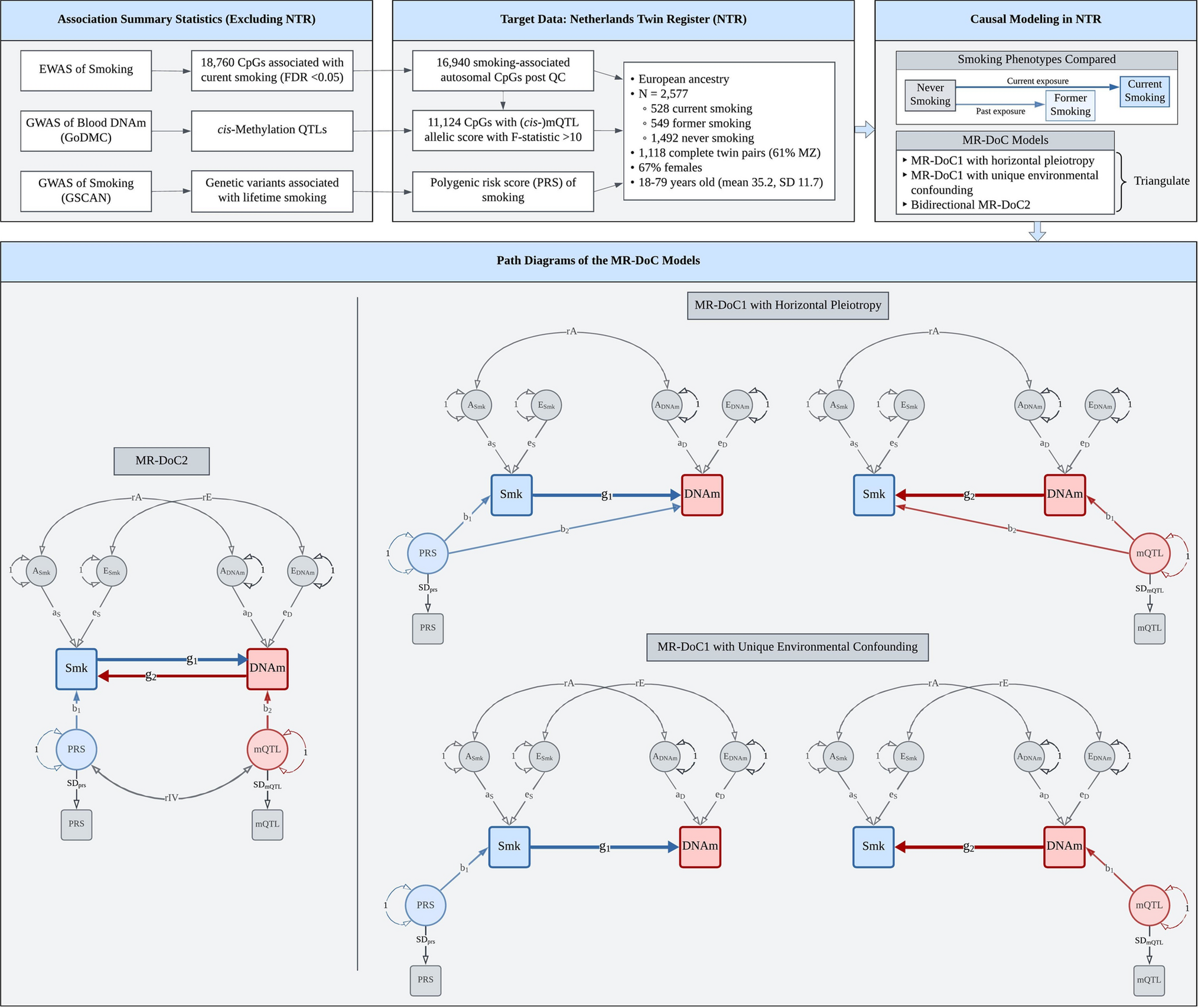

Our study leverages the nationwide database of the Netherlands Cancer Registry (NCR) with long-term follow-up linked to the Dutch Nationwide Pathology Databank (Palga), and samples collected from the pathology archives via the Dutch National Tissue Portal (DNTP). Cutaneous melanoma in the Netherlands is routinely registered by the Netherlands Cancer Registry (NCR). This registry is embedded in the Netherlands Comprehensive Cancer Organization. The NCR registers newly diagnosed malignancies upon automated notifications by Palga since 1989 [31].
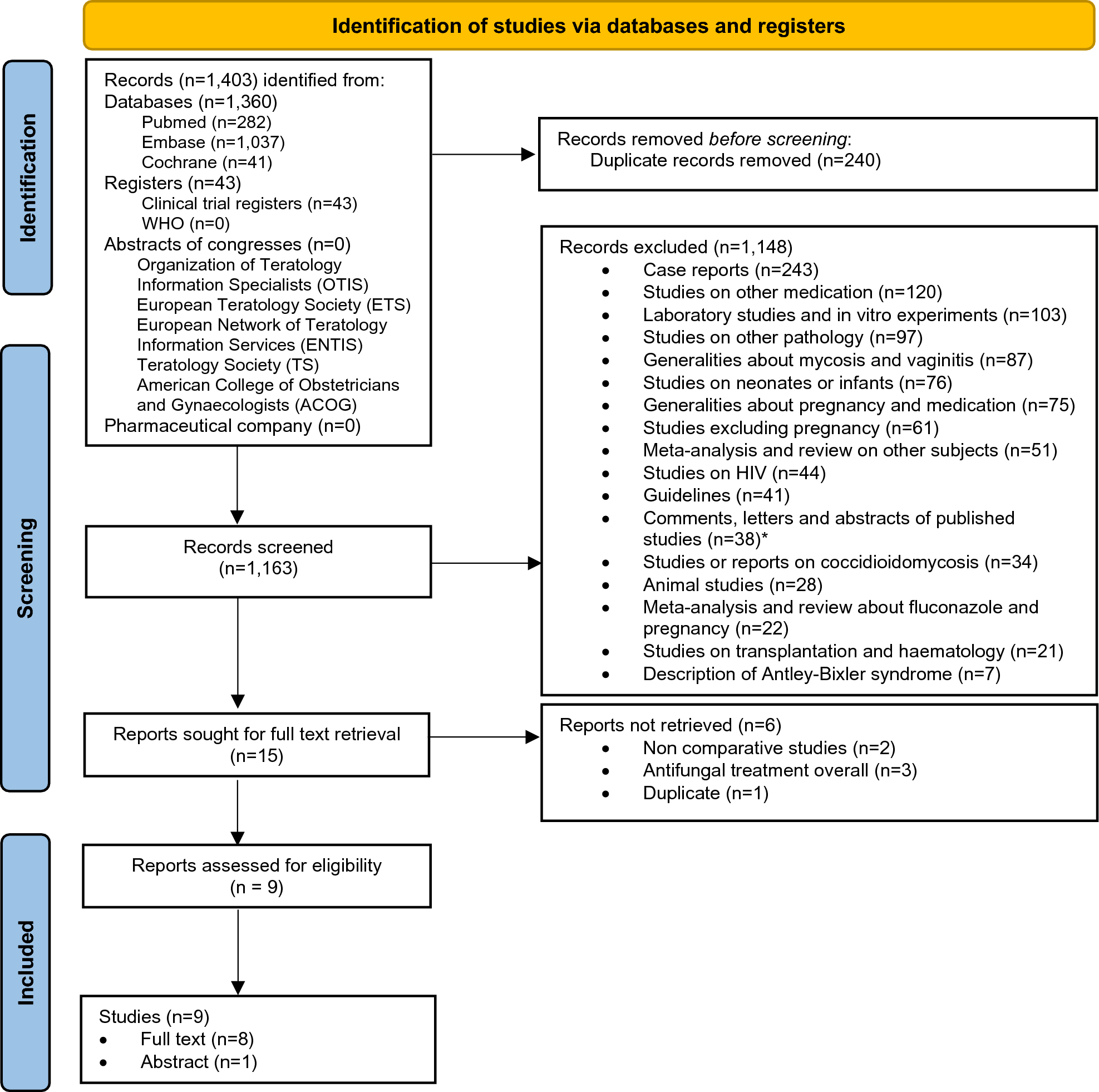
For this study, we used data on patients with cutaneous melanoma who were diagnosed with stage I or II disease at initial diagnosis. The retrieval was based on morphology codes M8720-8790 and topography code C44 of the third edition of the International Classification of Diseases for Oncology (ICD-O3) [32]. Melanomas of unknown primary site (C80.9) and patients with multiple melanomas were excluded. Trained data managers collected the data from pathology reports and digital patient records. We retrieved the following variables from the NCR: sex, age at diagnosis, year of diagnosis, vital status, topography, morphology, Breslow thickness, ulceration, clinical and pathological tumor-node-metastasis (TNM) stage, AJCC stage and SLNB. The AJCC stage used corresponded to the version valid at the time of diagnosis: AJCC 6 for 2009 and earlier, AJCC 7 for 2010–2017, and AJCC 8 from 2018 onward. Since July 2017, the NCR registers disease progression to stage III and IV during follow-up. For melanomas diagnosed before this date, we used data from Palga to obtain information about disease progression, including incidence dates and localizations of loco-regional (stage III) and distant recurrences (stage IV). Follow-up of recurrence and vital status of all patients in the study was updated until February 2024.
The registration of disease progression includes both patients with histopathologically confirmed and non-histopathologically confirmed metastases (e.g. observed with imaging, based on digital patient records) since 2017. Prior to this period, data were limited to cases with histopathologically confirmed metastases, potentially introducing selection bias. To address this, we expanded our patient selection to include individuals who, after initially being diagnosed with stage I or II disease, were later found to have distant metastases as per the digital patient records from the Erasmus MC Cancer Institute prior to 2017. Data and material were retrieved using linkages with aforementioned databases.
The Dutch Nationwide Pathology Databank (Palga) facilitates the use of nationwide histo- and cytopathology data by its decentralized information system, central databank and dedicated communication and information exchange infrastructure [33]. The registry was set up in 1971, under the name Palga (“Pathologisch Anatomisch Landelijk Geautomatiseerd Archief”). Nowadays, it encompasses all pathology laboratories in the Netherlands. Excerpts of the pathology reports are transferred to the central databank. Encrypted patient identifiers and demographic data are included. Based on the excerpts, a structured and coded Palga diagnosis is formed based on topography, morphology, function, procedure and disease.
The Dutch National Tissue Portal (DNTP) was used to request the selected tumor material. The DNTP utilizes Palga’s nationwide network and facilitates the use of tissue samples for research [34]. The DNTP consists of a centrally organized internet portal to request (tumor)tissue material. Dedicated employees provide practical support at peripheral pathology departments in the Netherlands.
The use of left-over diagnostic tissue samples for scientific research is based on the ‘no objection’ principle, as stated in the Code of Conduct for Health Research from the Committee on Regulation of Health Research. Consequently, a waiver of informed consent was granted [35].
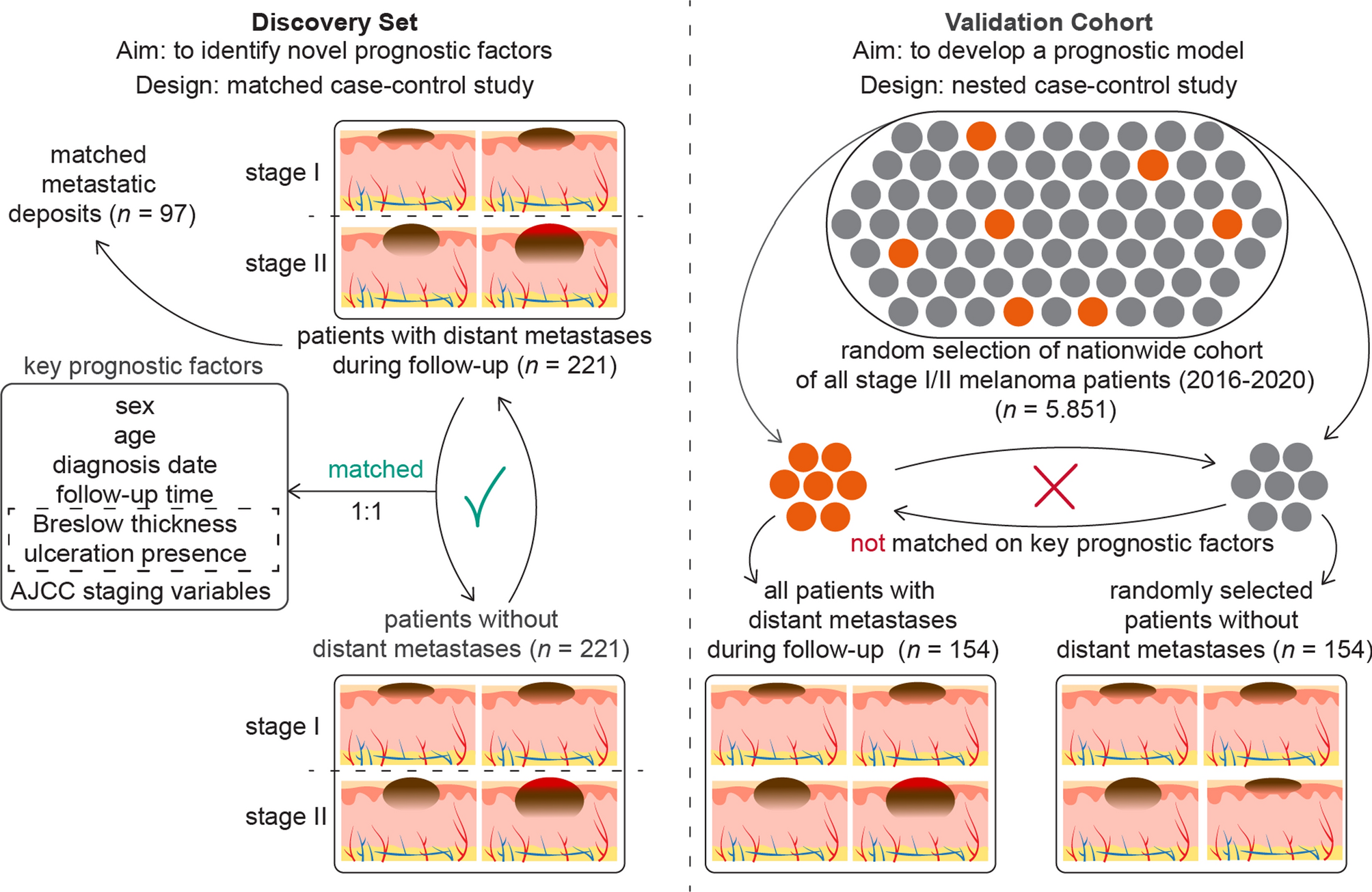
Study designsWe established a discovery set using a matched case–control design, and a validation cohort using a nested case–control design. In the discovery set, early-stage melanoma patients who developed distant metastasis during follow-up, were matched based on staging and well-known prognostic variables. We chose distant metastasis as the main outcome because these patients are at a higher risk of disease-specific death and represent the target group for systemic therapy among stage I/II melanoma patients. The aim of the matching approach is to identify novel prognostic factors that are independent of the already well-established prognostic factors.
To validate these novel factors and to build a prognostic model, we created a validation cohort structured as a nested case–control study derived from a comprehensive nationwide cohort. This validation cohort will be used to develop a clinical prediction model that incorporates both known and newly identified prognostic factors from the discovery set. If the final sample size of the validation cohort is sufficient, it may be divided into two parts: one for the development of the clinical prediction model and the other for its external validation. The nested case–control design enables the calculation of absolute risk probabilities associated with these novel factors, providing a measure more relevant than relative risk for clinical practice. Such a framework enhances the practicality and effectiveness of our prediction model in real-world clinical scenarios [36, 37]. A schematic overview of the study designs is provided in Fig. 1.
Fig. 1
Matched case–control design of the discovery set and nested case–control design of the validation cohort. (Abbreviations: AJCC = American Joint Committee of Cancer)
Discovery setA matched case–control study was conducted involving adult patients diagnosed with a single stage I or II melanoma. Patients who developed distant metastases during follow-up (i.e. cases) were matched with those who did not (i.e. controls). Matching criteria included the AJCC 8 staging variables—Breslow thickness and presence of ulceration—along with age at diagnosis and sex, due to their established prognostic significance [14,15,16,17].
To identify cases, we initially selected all adult patients who progressed from stage I and stage II disease to distant metastasis between 2013 and 2019 based on digital patient records from the Erasmus MC Cancer Institute. Given the relative infrequency of distant metastases following stage IA and IB diagnoses, we expanded our selection to include patients from the NCR, to supplement those from the Erasmus MC Cancer Institute. No restrictions were placed on the time to metastases for cases, thereby including both tumors that metastasized early and those that progressed later (e.g., 5–10 years post primary diagnosis). We aimed for an equal distribution of cases across stages I and II to facilitate stratified analyses. To ensure the most complete follow-up data, we requested updates on locoregional and distant disease progression from the NCR, along with the vital status of patients, in February 2024.
Matching on categorical variables was exact. For continuous variables, matching was categorized and prioritized as follows: Breslow thickness, age, and follow-up time. Specifically, a case diagnosed with distant metastases after x days was matched to a control whose follow-up time was equal to or greater than x days. Breslow thickness matching used intervals of 0.25 mm for up to 2.0 mm, 0.50 mm for 2.00–4.00 mm, and 1.00 mm for 4.00 to 10.00 mm, with no specific intervals for thicknesses greater than 10.00 mm. Age was matched within 5-year bands. An example of prioritizing matching variables could be illustrated by a 62-year-old case with a Breslow thickness of 1.4 mm being matched to a 67-year-old control who also has a Breslow thickness of 1.4 mm, instead of a 63-year-old control with a Breslow thickness of 1.3 mm.
After selecting cases and their matched controls, we linked the data from the NCR to Palga to obtain complete pathological histories for the selected patients. This included data on the primary melanoma and any histopathologically confirmed loco-regional or distant metastases. We manually reviewed pathology reports and excluded any patients with multiple melanomas, non-cutaneous melanomas, those initially diagnosed at stage III, or those with unclear pathology reports concerning their primary melanoma. Despite initially filtering for stage I and II patients with a single primary melanoma within the NCR, a few cases with multiple melanomas were included in the selection and subsequently excluded after reviewing the pathology data. Initially, cases were matched to controls at a 1:10 ratio to ensure that at least one suitable control could be selected from the pathology records and to provide flexibility in selecting a new control if the formalin-fixed paraffin-embedded (FFPE) tumor sample could not be retrieved. We ultimately selected one control per case for the retrieval of tumor samples and further analysis.
Validation cohortThe validation cohort comprised adult patients diagnosed with a single stage I or II melanoma between 2016 and 2020 in the NCR. We randomly selected a sample from this source cohort large enough to ensure a minimum of 400 eligible cases. These selected patients were then linked to Palga to retrieve complete pathological histories. From this pool, we identified cases and matched them to controls at a 1:5 ratio, ensuring the retrieval of FFPE tumor material for one control per case. Similar to the discovery set, pathology reports for all cases and potential controls were reviewed and selected. Based on the proportion of approved cases of the eligible ~ 400 cases, we calculated the size of the nationwide source cohort, which is a random sample from the nationwide stage I/II melanoma cohort.
Unlike the discovery set, we did not match the cases and on controls on age, sex, Breslow thickness and presence of ulceration, but did for follow-up time, as required for a nested case–control design to calculate absolute risks [36]. Additionally, based on differentially expressed genes identified during the quality control phase of the discovery set, we matched cases and controls on the type of surgical procedure (punch or shave biopsy vs. elliptical excision). For logistic efficiency, we also matched cases and controls based on the pathology lab of origin. This approach was not intended to address potential biases, as no such biases related to the pathology lab were observed in the RNA sequencing data from the discovery set. Each case was matched with one control for further processing. The retrieval and sectioning process of tumor specimens for the validation cohort mirrored that of the discovery set.
Tumor sample processingWe obtained FFPE tumor specimens, corresponding Hematoxylin & Eosin (H&E) slides, and anonymized pathology reports of the primary melanoma from all contributing pathology laboratories via the DNTP. To avoid the effects of tissue alterations from previous biopsies, we used tumor specimens from the initial surgical procedure of the primary melanomas.
From the H&E slides provided by the originating pathology laboratories, one FFPE block that best represented the tumor for further processing was selected. Each chosen block was then sectioned to produce a new H&E slide, which was subsequently digitalized. A dermatopathologist (A.M.) reviewed these digitalized slides to confirm the representativeness of the melanoma and to ensure the accuracy of the recorded Breslow thickness and ulceration status. If discrepancies were noted between our H&E slides and the original data, we reassessed the H&E slides from the original laboratory. If inconsistencies persisted and it was not feasible to select an alternative control, we excluded the set from further analysis.
To develop a prognostic model integrating multi-omics, imaging, and clinical data, we prepared slides from each sample for H&E staining, RNAseq, DNAseq, immunohistochemistry (IHC), and multiplex immunofluorescence (MxIF) from both the discovery set and the validation cohort (Fig. 2).
Fig. 2
Clinical, imaging, RNA sequencing and DNA sequencing data derived from the discovery set will be integrated and the most prognostic features will be validated in the validation cohort
We sectioned 4 µm slides from the selected FFPE blocks, alternating the sectioning order to ensure uniform representation of the entire tumor sample (Supplementary Information 1). To prevent contamination, a new blade was used for each section. Slides designated for IHC and MxIF were dipped in 60° Celsius paraffin to preserve tissue integrity. RNA was extracted from whole slides to include RNA from both tumor cells and surrounding immune cells, enhancing our analysis of the tumor microenvironment. RNA isolation was conducted as promptly as possible, within one week for most samples, to prevent degradation. For DNAseq, we performed macrodissection to predominantly obtain DNA from tumor cells. Given DNA’s greater stability, no strict timing requirements were applied before DNA isolation. Details regarding the RNA and DNA isolation and sequencing processes will be provided in separate manuscripts, as they fall outside the scope of this epidemiological design description.
In the discovery set, each FFPE block yielded 54 slides: 11 for H&E staining, 10 for DNAseq, 13 for RNAseq, 3 for IHC and 17 for MxIF. The first slide was always an H&E slide, which was digitalized to assess initial tumor representativeness and to record various histopathological variables. For each set of 10 MxIF slides, an adjacent H&E slide provided a morphological reference, facilitating more precise analyses. Similarly, from the validation cohort, we processed 53 slides per FFPE block: 10 for H&E, 10 for DNAseq, 13 for RNAseq, and 20 for IHC or MxIF, with the first 10 MxIF/IHC slides accompanied by an adjacent H&E slide (Supplementary Information 1).
To explore the clonal heterogeneity between primary melanomas and their corresponding distant metastatic deposits, we identified all histopathologically and cytopathologically confirmed distant metastases, as recorded by Palga, utilizing the detailed pathological histories available to us from the discovery set. Following the procedure established for primary tumors, we procured FFPE blocks and cytological slides from the DNTP. For each sample, the H&E slide was reviewed by a pathologist (A.M.) to confirm the presence of at least 20% tumor area, and 10 slides were generated for DNA isolation. Investigating the genetic relationships between primary tumors and their metastases will help in identifying and excluding cases where the distant metastases may not be directly derived from the identified primary melanoma, suggesting the presence of another, unknown primary tumor.
Statistical analysesThe sample size for our discovery set was derived from the requirements of developing an RNA gene expression profile. Lacking specific features and effect sizes, we referenced a previous study that successfully developed a gene expression profile to predict low risk of SLN metastasis among melanoma patients [38]. In this previous study, differential gene expression analyses were conducted on 6 samples, which led to identifying 54 candidate genes tested across 160 patients. To enhance our capability to study all expressed genes comprehensively, we doubled the initial sample size from the referenced study, aiming for at least 350 patients, with 175 patients who developed distant metastases and 175 who did not. This sample size ensures at least 80% power to detect prognostic factors with an odds ratio (OR) of ≥ 2 occurring at a frequency of 20%, or an OR of ≥ 3 for factors occurring at a frequency of 5%.
The validation cohort comprises a sufficient number of events for robust model development, as this determines the effective sample size. Although various thresholds exist for the number of events per variable in different contexts, at least 10 events per variable are generally recommended for accurate prediction modeling of binary outcomes [39, 40]. Achieving this ratio requires shrinkage of the regression coefficients in the final model to prevent overfitting. Therefore, we aimed to include at least 100 cases and 100 controls in the nested case–control design of the validation cohort. With 10 events per variable, we can allocate 10 degrees of freedom in a regression model, which is adequate to incorporate known predictors (age, sex, Breslow thickness, and ulceration) along with novel prognostic factors identified from a pre-trained model of the discovery set.
We intentionally oversampled the initial validation cohort and requested a higher number of FFPE tumor specimens to ensure that a sufficient sample size was achieved within project timelines. Upon receiving and evaluating the available cases, we adjusted the size of the nationwide source cohort proportionally based on the percentage of cases received—for instance, if only 50% of the expected cases were obtained, we then randomly sampled 50% from the baseline cohort.
The descriptive statistics of the distribution of the clinical characteristics for both the discovery set, the validation cohort and the nationwide source cohort are presented. Differences in age, sex, Breslow thickness, presence of ulceration, AJCC stage, body site of the primary melanoma, morphological subtype and performed SLNB’s between cases and controls were tested. Categorical variables were analyzed using the McNemar’s test or the McNemar-Bowker test for > 2 categories, and continuous variables were assessed using the Wilcoxon signed-rank test, all accounting for the paired nature of the data. The level of statistical significance was set at a two-sided p < 0.05.
Statistical analyses were conducted using SAS® (9.4 M8), R Studio (version 4.3.3), and IBM® SPSS® software (version 29.0).
Comments (0)