
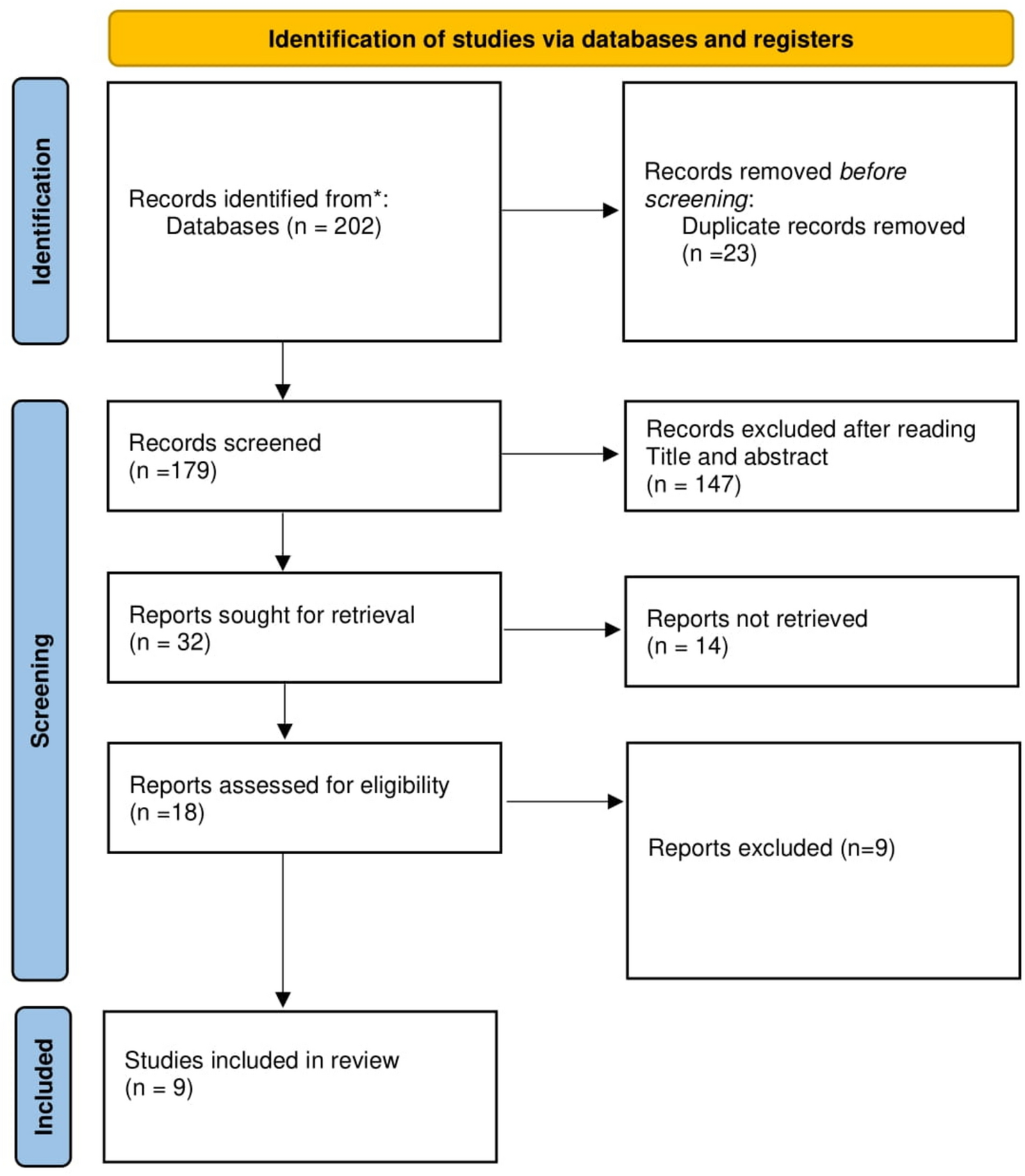
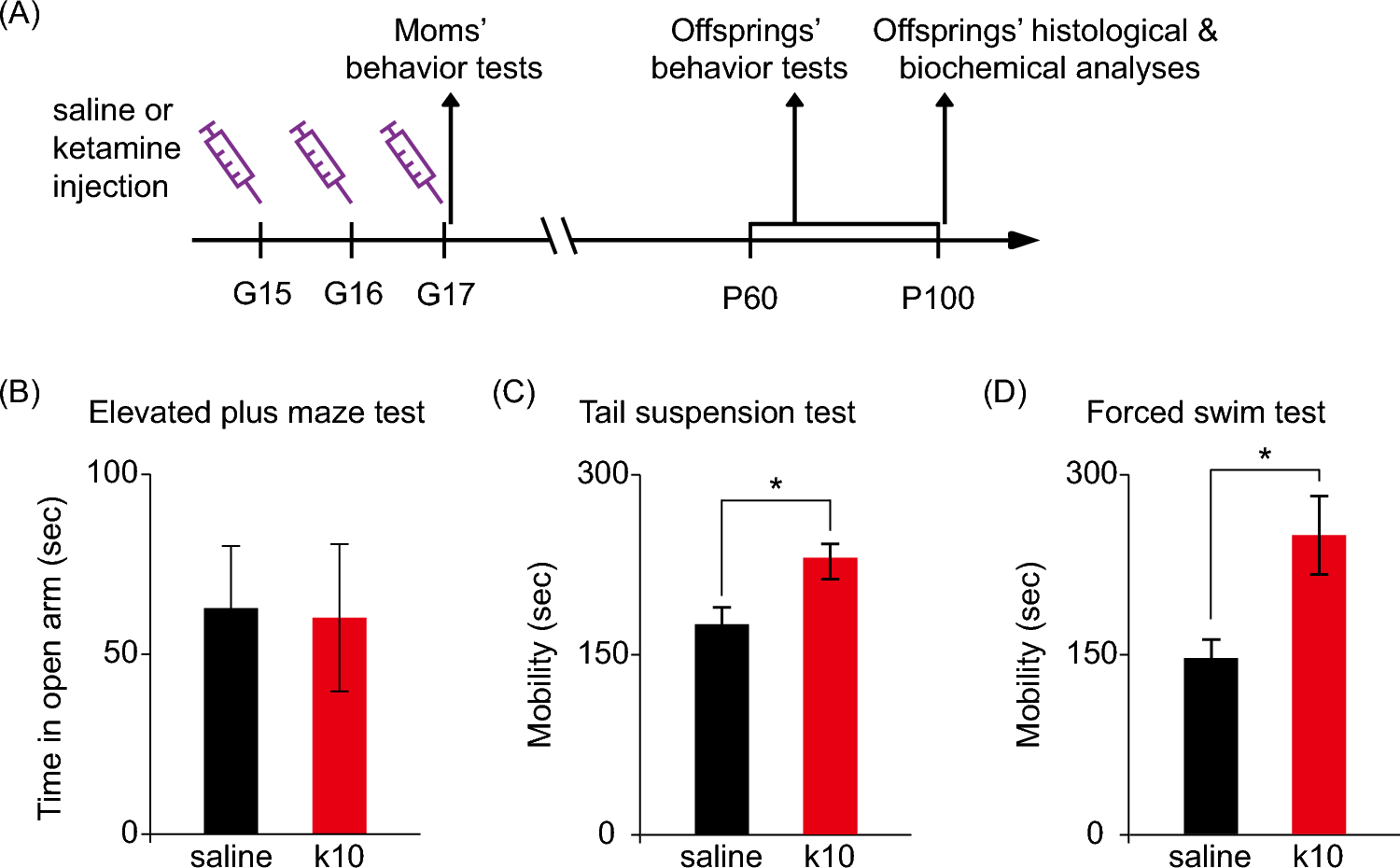
Remember me



To investigate the immediate events following a contusive spinal cord injury, a custom-made impactor was employed to induce a physical injury at thoracic spinal cord level of an ex vivo preparation of entire CNS (Mohammadshirazi et al. 2023). The careful design of the impactor included a proper shielding to minimize any electrical interference during operation, to allow simultaneous electrophysiological recordings during the impact. In an exemplar experiment, a brief and intense impact (duration = 1.30 s, displacement = 2656 µm) on the ventral cord (T10) led to a massive depolarization, recorded rostral and caudal to the compression site from cervical and lumbar VRs, respectively (Fig. 1A, B). The profile of the average injury-induced potential from VRrL5 reveals a peak of 8.21 ± 1.32 mV and a latency of 178.41 ± 15.17 ms after the impact, recovering to 81.11 ± 12.56% 6 min later (n = 5; Fig. 1G). A milder impact (displacement = 625 µm) applied to another group of five preparations elicited a smaller injury-induced potential from VRrL5 (SI. Fig. 1A), with a peak of 0.76 ± 0.38 mV (SI. Fig. 1B) and a latency of 180.84 ± 24.61 ms (SI. Fig. 1C), which recovered to baseline just 2 min later.
Fig. 1
A transient depolarization immediately follows a physical injury to the spinal cord. A Long continuous recordings from VRrL5 and VRrC2, while the cord is being impacted at T10 (red arrows). An injury-induced potential occurs after 194.4 ms from the onset of the impact on VRrL5, and after 225.2 ms on VRrC2. On VRrL5, a depolarization peak of 6.86 mV is reached after 2.66 s, followed by a depolarizing plateau lasting 3.52 s and spontaneously recovering to baseline in less than 15 min. VRrC2 generated a smaller depolarization peak (1.47 mV). Before and after the impact, 10 mM KCl were perfused for ten minutes to compare the recruitment of motor pools. Before the impact, KCl generated depolarizations that were smaller on VRrL5 (40.34%) and greater on VRrC2 (177.9%), compared to the ones induced by the following impact. A second exposure to 10 mM KCl after injury produced on both VRs the same depolarizations as the pre-impact application. B Magnification highlights the depolarization at VRrL5 in the first five seconds after impact (red arrows). C and D Faster time scales of VR traces in A, corresponding to the shaded blue and green fields that are recorded before and after the impact, respectively. After the impact, spontaneous sporadic bursting from VRs is largely reduced by the trauma. E Pooled data from five experiments, displays an amplitude of impact-induced depolarization recorded from VRrL5 that significantly exceeds the depolarization-induced by 10 mM KCl before and after the impact (*P < 0.001, repeated measures ANOVA followed by Dunnett all pairwise multiple comparisons test, n = 5). Mean values are indicated by the red dots and line. F In cervical motor pools, depolarization after injury is notably smaller than after a second application of potassium (*P = 0.046, repeated measures ANOVA followed by Dunnett all pairwise multiple comparisons test, n = 5). Mean values are indicated by the red dots and line. G Superimposed depolarizations from VRrL5 in five experiments. H SMI 32 labeling of samples collected 90 min after the impact display a comparable number of motoneurons in L1 to L3, and in L3 to L5 segments of both, intact and SCI experiments. I Box-and-whisker plots for the number of SMI 32 positive cells normalized to the total number of nuclei in the region of interest (ROI) show no statistical changes caudal to the injury site (L1–L3 and L3–L5, P = 0.360, ANOVA followed by Tukey–Kramer all pairwise multiple comparisons test, n = 4 intact, n = 4 SCI)
Sporadic episodes of spontaneous motor discharges appear synchronous among all motor pools of the isolated neonatal spinal cord as a result of the motor activity reverberating through a diffuse propriospinal network (Cazalets 2005). This activity was largely reduced on both VRrL5 and VRrC2, following an impact at T10 (Fig. 1C, D), as observed in 20 out of 24 preparations.
Each spinal segment owns a distinct number of motoneurons (Sadeghinezhad and Nyengaard 2021) that varies the absolute magnitude of each segmental motor pool recruitment. In addition, the amplitude of extracellular signals depends on the impedance of glass electrodes, which is mainly affected by the seal of the target spinal root. To quantify the peak of injury-induced depolarization in relation to the depolarization produced by the direct recruitment of motoneurons in each spinal segment and across different preparations, 10 mM potassium was applied to the bath for 10 min before the impact to the same exemplar preparation (Fig. 1A). A second exposure to 10 mM KCl was applied to ascertain the absence of any functional alterations of motor pools after the impact (Fig. 1A). Pooled data from five preparations showed that the peak of average injury-induced depolarizations from VRrL5 was significantly higher than the depolarizations elicited by 10 mM KCl (P < 0.001, repeated measures ANOVA followed by Dunnett all pairwise multiple comparisons test, n = 5; Fig. 1E). Conversely, in the same group of preparations, the average injury-induced depolarization from VRrC2 was lower than the one elicited at lumbar levels (P < 0.001), and significantly lower than the depolarization determined by a second application of 10 mM KCl (Fig. 1F; P = 0.046, repeated measures ANOVA followed by Dunnett all pairwise multiple comparisons test, n = 5). Notably, at both L5 and C2 levels, potentials elicited by rising KCl concentrations were comparable before and after the impact (Fig. 1E, F). This confirms that an injury targeted to the low thoracic cord (T10) does not reduce the overall availability of motoneurons located in motor pools far from the injury site, which remain equally functional once directly activated by KCl.
Furthermore, distinct lumbar segments of intact and injured spinal cords were treated with a selective marker for motoneurons in the ventral horns (SMI 32 antibody). Histological processing visualized a similar SMI 32 staining in the ventral cord of the intact and injured preparations, for both L1-L3 and L3-L5 segments (Fig. 1H). Mean data from 49 tissue sections from a total of eight animals (four intact and four injured spinal cords) confirmed no significant difference in the ratio of SMI 32 positive cells (Fig. 1I; P = 0.360, ANOVA followed by Tukey–Kramer all pairwise multiple comparison test), hence excluding the acute death of any lumbar motoneurons after the low thoracic injury and related spread depolarization.
Collectively, a physical insult to the mid-thoracic spinal cord triggers a transient and massive depolarization spreading along the entire spinal cord, suppressing the spontaneous motor activity that is derived synchronous among all neonatal VRs, yet without any cellular loss of lumbar motor pools.
Injury Potentials Originate from Actual Neuronal DepolarizationsTo confirm that the observed sudden increment in DC levels is indeed a genuine potential rather than an artifact, we performed supplementary tests. Firstly, in five experiments, repeated impacts of increasing strength on the same preparations (SI. Fig. 1) demonstrated that stronger impacts (625 µm, 1250 µm, 1875 µm) produce higher potentials. However, an additional increase in the intensity of the trauma (2656 µm) failed to further increase the amplitude of injury potentials, likely due to the repetitive damage to the cord at the same site of impact (SI. Fig. 1). Overall, any electrical interference produced by the engine of the device remained equal whenever the device was activated, regardless the extent of rod displacement. Conversely, the increasing potentials obtained in the present study in response to greater impact strengths prove the direct relationship between the severity of impact and the extent of VR depolarization.
Another test considered the device acting solely on the bath, close to the preparation, but without touching the cord (SI. Fig. 3A, B), revealing the absence of any potentials generated by the impactor engine. Furthermore, in four experiments, multiple impacts of equal severity (displacement = 2656 µm) were serially applied to the same site (T10) for five times, with a lag of less than 10 s between any two consecutive impacts. As a result, peaks of injury-induced potentials remained stable, excluding any summation of artifacts (SI. Fig. 3C). On the other hand, when the impact was delivered at the top of a large depolarization (16.46 mV) that was evoked by 50 mM KCl, no injury-evoked depolarization appeared (SI. Fig. 3D). Finally, in another preparation, no baseline deflections were recorded from VRrL5 when the impact was inflicted to the T10 segment of a spinal tissue inactivated by both high temperature (100 °C) and long-lasting (1 h) oxygen deprivation (SI. Fig. 2C), proving the biological origin of depolarization after injury.
Collectively, these tests revealed the absence of any significant baseline drift produced either by the engine itself or by the sudden movement of the tip in the recording bath.
Injury Potentials Propagate Rostrally and Caudally from the Site of Impact in Ventro-Dorsal DirectionsTo better investigate the propagation of injury-induced depolarization along the entire spinal cord, we collected data from numerous VRs, out of a dataset of 44 preparations injured at the ventral aspect of T10 with the strongest impact (2656 µm tip displacement, Fig. 2A). Injury potentials of different amplitude were recorded from distinct spinal segments, with the highest peaks from VRL1 and L2 being significantly larger than those derived at the extremities (Fig. 2B, see Table 1 for statistical details). Injury potentials progressively slowed down the farther they were recorded from the impact site, with the lowest latency recorded at VRL1 (Fig. 2C, see Table 2 for statistical details). Resulting velocity of the rostro-caudal conduction of injury-induced depolarizations from the site of impact to VRL1 (4.44 mm far from impact) was 0.03 ± 0.01 m/s, equal to the caudo-rostral conduction from the site of impact to VRT5 (4.83 mm far from impact, P = 0.451, Mann–Whitney test, n = 3 for T5 and n = 18 for L1).
Fig. 2
Impact-induced depolarization spreads from the injury site to the whole spinal cord. A A ventral view of the CNS preparation with dorsal vertebrae attached. VRs recordings are taken from the VRs indicated by dotted yellow lines, while the injury site at the T10 segment is highlighted by a red dot. B Mean amplitudes of injury potentials from several VRs. Red dotted line indicates the level of injury (T10). Number of experiments for each VR is indicated in brackets. Statistically significant amplitudes are indicated by *, as described in Table 1. C Mean latencies of injury potentials from several VRs. Red dotted line indicates the level of injury (T10). Number of experiments for each VR is indicated in brackets. Statistically significant amplitudes are indicated by *, as described in Table 2. D Superimposed mean traces from simultaneous recordings of injury potentials from both, DR (green trace) and VR (blue trace), at L1 (n = 4). E and F Injury potentials from DRrL1 are significantly smaller (E; *P = 0.041, paired t-test, n = 5) and slower (F; *P = 0.015, paired t-test, n = 4) than recorded from VRrL1. Red dots and line show average values
Table 1 Amplitude values of impact-induced depolarizations from different VRsTable 2 Latency values of impact-induced depolarizations from different VRsTo gain insights on the dorsal–ventral propagation of injury-induced depolarization, we simultaneously derived from both VRrL1 and DRrL1 while impacting the ventral side of the cord at T10. Data pooled from many experiments (Fig. 2D) indicates that the impact leads to injury potentials that propagate also to the dorsal part of the cord, although they appear smaller (Fig. 2E; P = 0.041, paired t-test, n = 5) and spread more slowly (Fig. 2F; P = 0.015, paired t-test, n = 4) than ventrally elicited potentials.
Present data indicates that a physical injury to the spinal cord elicits a strong wave of depolarization that departs from the site of injury and invests the entire spinal cord with the same velocity, affecting also dorsal segments. This observation provides the rationale for ascertaining the functionality of spinal networks above and below the site of injury.
An Impact Generates Potentials that Equally Propagate to Both Sides of the Cord, and Functionally Disconnects the Lumbar Cord from Descending Respiratory InputTo confirm the symmetrical propagation of injury-induced depolarizations along both sides of the cord, simultaneous VR recordings were obtained from both left and right VRs at L1, in response to a physical injury at T10. In a sample experiment, continuous recordings were acquired from VRlL1, VRrL1, and VRrC2 (Fig. 3A). Average data from four experiments indicated an equal extent of impact-induced depolarizations on both sides of the L1 spinal segment (Fig. 3B, P > 0.999, Wilcoxon matched-pairs signed-ranks test, n = 4).
Fig. 3
Impact evokes equal bilateral injury potentials and disconnects lumbar motor pools from descending respiratory input. A Continuous and simultaneous recordings from VRrL1, VRlL1, and VRrC2 showing the exposure to 10 mM potassium (10 min) and to the following impact at T10. After the impact, VR injury-induced potentials peaked at 10.15 mV and 11.38 mV for left and right VRs, respectively. B The plot visualizes the equal amplitude of injury-induced depolarizations recorded from left and right L1 VRs (P > 0.999, Wilcoxon matched-pairs signed-ranks test, n = 4). Red dots and red line correspond to average values. C Magnifications correspond to the pale regions of continuous traces in A, and highlight rhythmic respiratory bursts in control (blue panel, 0.02 ± 0.01 Hz) and 21.8 min after the impact (green panel, 0.02 ± 0.01 Hz). Fictive respiration originating from brainstem structures is maintained at VRrC2 but disappeared from lumbar VRs due to the functional interruption of descending input beyond the site of impact
To monitor the respiration-related activity that originates by neuronal networks located in the brainstem (Del Negro et al. 2018), spontaneous rhythmic bursts were recorded from cervical VRs of the isolated CNS (Nicholls et al. 1990; Iizuka 1999; Mohammadshirazi et al. 2023; Apicella and Taccola 2023). To assess any early and transient alteration of the respiratory rhythm during the impact, 20 respiratory bursts from cervical VRs were analyzed right before and soon after the injury. In 4 out of 7 preparations, the first respiratory event after the impact was delayed, showing an early perturbation of the neuronal networks in the brainstem generating the respiratory rhythm (SI. Fig. 4). Albeit not consistent among all preparations, this effect was observed in the majority of experiments (57%), regardless of the magnitude of injury potentials from cervical VRs and the age of animals (SI. Fig. 4). Noteworthy, when assessed 30 min after injury, respiratory bursting recorded from upper cervical VRs, was not affected by the thoracic impact to the cord (Fig. 3C). In seven preparations, respiration frequency from VRC2 was 84.28 ± 20.29% of pre-impact control (0.05 ± 0.03 Hz from 20 min pre-injury, 0.05 ± 0.02 Hz from 20 min post-injury, P = 0.709, paired t-test) with unaffected burst amplitude (0.29 ± 0.18 mV before impact, 0.26 ± 0.21 mV after impact; P = 0.195, paired t test). Moreover, the respiratory rhythm can also be recorded from lumbar VRs, which drive the recruitment of chest muscles to assist the expiratory phase (Janczewski et al. 2002; Taccola et al. 2007; Giraudin et al. 2008). In seven experiments, the lumbar respiratory motor activity is abolished after trauma (Fig. 3 C).
In summary, the equal magnitude of bilateral injury potentials propagating to lumbar VRs confirms the midline location of the impact. Moreover, the disappearance of respiratory bursts below the site of injury indicates that lumbar motor pools are completely disconnected from supraspinal respiratory centers.
Impact Causes Extensive Neuronal Loss at the Contusion Site and Functionally Disconnects Ascending Afferent InputDisappearance of respiratory episodes from the lumbar cord indicates that descending respiratory input from the brainstem are blocked at the level of impact. To investigate whether also the conduction of ascending input is blocked by the impact, we recorded ascending input evoked by continuous electric stimulations (intensity = 100 µA, pulse duration = 0.1 ms, frequency = 0.1 Hz) of sacrocaudal afferents (Etlin et al. 2010). Simultaneous recordings were taken above and below the level of impact. In a sample experiment, single reflex responses in control were recorded from VRrL5 and VRrC2, respectively (blue traces in Fig. 4A). At the peak of injury-induced depolarization, both responses vanished (Fig. 4A). After 38 s from the impact, reflex responses from VRrL5 reappeared and eventually stabilized after 8 min, albeit reduced in amplitude to 41% of pre-impact control. Contrariwise, cervical responses were completely abolished (green traces in Fig. 4A). The disappearance of cervical reflexes after the impact was replicated in nine out of nine preparations.
Fig. 4
Contusion suppresses ascending conduction of afferent input and causes massive neuronal death at the site of ventral impact. A The cartoon depicts the CNS preparation with the impact site on the ventral aspect of T10 (red dot). Extracellular electrodes are positioned at C2 and L5 rVRs, and repetitive electrical pulses (0.1 Hz, 100 µA, duration = 100 µs) are supplied to sacrocaudal to elicit ascending input (arrow). Right traces show simultaneous recordings from VRrL5 and VRrC2 with reflex responses appearing in control and magnified in the blue insert. After the depolarization induced by the impact (red arrow), evoked motor responses are abolished on both VRs. During repolarization, responses progressively reappear on lumbar VR, while lumbar reflexes become visible again after 38 s from the impact and recover towards the original size by the time (8 min, top green insert). Contrariwise, reflexes from VRrC2 do not recover (bottom green insert). B Reconstruction of sagittal tissue sections of a spinal cord (caudal left, rostral right, ventral up, dorsal down) as processed with DAPI and NeuN staining. A massive cellular loss is visible on the ventral aspect of the impact site. The base of the dotted yellow rectangle centered at T10, is calibrated to the width of the impactor tip. C Magnifications of sagittal tissue sections as in B stained with DAPI and NeuN, and collected from serial spinal segments at caudal level (T11-L1, left), injury site (T9-T11, centered at T10, middle) and rostral spinal cord (T9-T6, right). The lack of NeuN (green) staining at the site of impact indicates extensive neuronal loss. D The plot quantifies the statistical reduction of NeuN-positive cells at the injury site compared to both rostral and caudal segments (*P < 0.001, ANOVA followed by Tukey–Kramer all pairwise multiple comparisons test, n = 5). Cell count is normalized to the total number of nuclei in the region of interest (ROI)
The suppression of both, respiratory lumbar episodes and cervical reflexes evoked by sacrocaudal stimulation, suggests a functional impairment of input conduction along the cord due to the impact. To visualize neuronal cell death caused by the impact, staining for neurons was performed on sagittal sections of the entire spinal cord. The ventral spinal cord at the site of impact (dotted yellow rectangle) showed negligible neuronal labeling for NeuN due to an extensive cell loss (Fig. 4B). In another example, magnifications of sagittal tissue sections from serial close spinal segments confirmed a lower number of NeuN positive cells at the injury site (Fig. 4C). Pooled data from five experiments demonstrated the significant reduction of NeuN-positive cells at the injury site (T9–T11, centered at T10) compared to rostral (T9–T6) and caudal (T11–L1) segments (P < 0.001, ANOVA followed by Tukey–Kramer all pairwise multiple comparisons test; see Fig. 4D).
This histological evidence describes a massive neuronal damage at the site of injury and proves the blockage at the impact site of electric signals that would have otherwise travelled along the spinal cord.
Spinal Cord Oxygenation Drops After a Spinal ImpactAfter an SCI, systemic hypotension and pericyte constriction of spinal capillaries decrease spinal oxygen delivery, reducing oxygen concentration on spinal tissues (Partida et al. 2016; Li et al. 2017; Williams et al. 2020). To quantify spinal cord oxygenation (SCO2) during contusion, an intraparenchymal probe for O2 was positioned 100 µm deep into the cord on the ventral funiculus between L1 and L2 VRs, while continuous electrophysiological signals were derived from VRrL1. The time course of average PO2 from nine preparations indicated that SCO2 in control (31.19 ± 7.36 Torr) dropped to 11.68 ± 4.03 Torr after the impact, and then slowly recovered to the 78.74% of control after 30 min (Fig. 5C).
Fig. 5
Impact drops SCO2 with a pattern that mirrors the profile of injury-induced depolarization. A Continuous trace from VRrL1 with a large depolarization at the site of impact at T10 (red arrow, 5.23 mV), which recovered to baseline after 12 min. B Simultaneous SCO2 measurements performed from the ventral funiculus between L1/L2 VRs in the same experiment as in A, with PO2 values oscillating between 20.44 and 50.91 Torr in control. PO2 drops right after the impact (8.12 Torr), eventually recovering to baseline after 10 min, mirroring the profile of DC level changes in A. C Average spinal PO2 profile before and after the impact (red arrow, n = 9)
SCO2 for ex vivo preparations parallels the level of cellular activity as the induction of rhythmic locomotor-like activity corresponded to a fall in tissue PO2. (Wilson et al. 2003). To provide a reference for SCO2 during a large depolarization, the CNS was perfused for 10 min with a modified Krebs solution containing 10 mM KCl. Potassium induced a mean depolarization of 1.83 ± 0.54 mV from VRL1, while average PO2 measured from the L1 spinal segment dropped to 9.54 ± 2.14 Torr (SI. Fig. 2A).
The link between the increased neural activity induced by a large depolarization and the PO2 drop was further explored using a CNS preparation that underwent a functional inactivation through heat-shock (100 °C) and then a continuous perfusion with oxygenated Krebs. Here, no depolarization was recorded from VRrL5 after exposure to potassium (10 mM), while an intraparenchymal probes for O2 inserted at L1 spinal level derived a mean PO2 of 528 ± 8.74 Torr equal to pre-K+ control values. In the same preparation, the spinal impact did not elicit any depolarizations from VRrL5, with PO2 measurements that remained unchanged before and during the impact (505.76 ± 2.57 in control and 508.75 ± 3.16 Torr during impact, SI. Fig. 2C).
Collectively, the impact induced a drop in SCO2 that mirrors the kinetics of impact-induced depolarization and was comparable to the activation of spinal networks following 10 mM K+.
Impact Transiently Suppresses Lumbar Motor ReflexesA compression of the spinal cord is followed by a spinal shock, characterized by the suppression of motor evoked responses below injury, lasting beyond the moment of the first insult (Ditunno et al. 2004). To confirm the presence of a shock phase in our ex vivo SCI model, stimuli were continuously supplied to sacrocaudal afferents (frequency = 0.1 Hz, intensity = 1.6–6.15 Th, pulse duration = 0.1 ms) while motor reflexes were derived from VRrL5 in control (peak amplitude = 0.77 ± 0.2 mV). The profile representing changes in the amplitude of reflex responses throughout the experiment displays a complete suppression of motor reflexes in correspondence to a localized impact at T10 (red arrow, Fig. 6A, B). The transient disappearance of lumbar motor reflexes after trauma at T10 was obtained in five preparations where reflexes were also halted at the early peak of the depolarization evoked by 10 mM K+. After 27.91 ± 6.06 s from the impact, smaller electrically evoked responses reappeared, and recovered to 90% of pre-impact values after 18.25 ± 12.2 min. In the same five preparations, the time of reappearance of the first reflex after impact was not correlated to the amplitude of the injury potential (correlation coefficient = − 443, P = 0.455). Through multiple simultaneous recordings, comparable transient suppressions of motor reflexes were reported across VRs at spinal segments L1, L2, L4, L5, L6 on both sides. To exclude that the transient halt of lumbar reflexes is due to an interference produced by the impactor movement, in one sample, lumbar responses were allowed to recover after being transiently abolished by a first impact at T10. Then, the spinal cord was completely transected at L1 level (SI. Fig. 5A, B) and, after at least 15/20 min, a second impact at T10 was performed, which did not evoke any injury potentials from the disconnected caudal cord nor varied the amplitude of reflex responses (SI. Fig. 5B). Noteworthy, the second impact still elicited an injury potential from the rostral cord (SI. Fig. 5B).
Fig. 6
Motor reflexes vanish at the peak of both, chemically- and impact-induced depolarizations. A A 178 min long recording from VRrL5 during the continuous delivery of electrical pulses to sacrocaudal afferents (frequency = 0.1 Hz; intensity = 100 µA, 5 × Th; pulse duration = 0.1 ms) to evoke motor responses. Motor reflexes are traced in control, during 10 mM K+ perfusion, wash out, impact to T10 (red arrow) and a second K+ application. The first exposure to K+ elicits an early depolarization of 4.05 mV and suppresses reflexes, which recover to baseline during washout in normal Krebs. Afterwards, at the peak of the impact-induced depolarization (9.69 mV), motor evoked responses are transiently suppressed but fully recover, as the baseline repolarizes, after 31.06 min from the impact. A second exposure to 10 mM K+ evokes an early depolarization of 4.55 mV, which abolishes motor reflexes until their full recovery during washout. Top inserts magnify single reflexes (dotted vertical lines correspond to artifacts of stimulation) for distinct instants of the experiment as indicated by the colored dots below. At the top of each depolarization, motor reflexes are suppressed (brown, red and purple top traces). B Time course of reflex amplitude for the trace in A demonstrates that reflexes vanish (amplitude = 0 mV) during sudden depolarizations elicited by either perfusing the entire CNS with potassium or by applying a localized impact to the cord
In summary, in the current study, the calibrated and localized impact to the cord has always been followed by a transient suppression of evoked reflexes from spinal motor pools.
A Thoracic Impact Alters electrically induced Fictive Locomotor PatternsResults collected so far indicate that, after an impact, the entire spinal cord experiences a transient large depolarization, with neuronal death only at the injury site. To explore whether the depolarization induced by the impact affects the functionality of lumbar spinal networks for locomotion, stereotyped trains of rectangular pulses (frequency = 2 Hz, intensity = 1–5 × Th, pulse duration = 0.1 ms) were applied to sacrocaudal afferents for 80 s (Fig. 7 A). In response to stimulation, episodes of locomotor-like oscillations were recorded in control and at different time points after the impact (15, 60, and 120 min post-SCI, Fig. 7A, B). Signals characteristically appeared double alternating between flexor-related signals from VRL2 and extensor-related commands from VRL5, as well as between left and right motor pools (Kiehn and Kjaerulff 1996). Pooled data from seven experiments showed that the impact unaltered several characteristics of fictive locomotion (SI. Fig. 6) but did reduce cumulative depolarization (Fig. 8A; P = 0.002, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 6) and amplitude of cycles from VRrL2 (Fig. 8B; P < 0.001, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 6). In addition, duration of fictive locomotion episodes from VRrL2 after 60 min from the impact (Fig. 8C; P = 0.031, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7), and period of cycles of VRrL2 after 15 and 60 min from the impact, were significantly lower than in control (Fig. 8D; P = 0.008, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7). Similarly, 15- and 60-min post-impact, episodes from VRrL5 were faster than in the control group (Fig. 8E; n = 6, P = 0.002, Friedman test followed by Dunn's multiple comparisons test vs ctrl), as well as more irregular at 15 min post-impact (Fig. 8F; P = 0.006, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7). Notably, after injury, oscillations from both extensor and flexor commands (Fig. 8G; homolateral CCF, P = 0.013, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7), as well as from the left and right sides of the cord (Fig. 8H; homosegmental CCF, P = 0.001, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7) exhibited poorer alternating coupling than controls.
Fig. 7
electrically induced fictive locomotion is affected by a localized thoracic compression. A Serial 2 Hz trains of stereotyped rectangular pulses (intensity = 125 µA, duration = 0.1 ms) are applied to sacrocaudal afferents to evoke epochs of locomotor-like oscillations from VRlL2, VRrL2, and VRrL5. Fictive locomotion patterns were recorded in control, and then 15 min, one hour, and two hours after injury. Fictive locomotor patterns recorded in control from VRrL2 were characterized by a cumulative depolarization of 0.7 mV with 28 superimposed alternating cycles (homolateral CCF = − 0.70, homosegmental CCF = − 0.87), defined by a peak amplitude of 0.33 ± 0.08 µV and a period of 2.89 ± 0.74 s. In the same preparation, impact reduced cumulative depolarization (0.5 mV, 15 min post-SCI), generating smaller (0.16 ± 0.06 µV, 15 min post-SCI) and slightly less regular locomotor-like oscillations (period CV = 0.28, 15 min post-SCI Vs. period CV in control = 0.26), regardless their unchanged number (28, 15 min post-SCI). B Magnifications of simultaneous traces (blue fields for VRlL2, green for VRrL2, and orange for VRrL5) correspond to oscillations captured at steady state in A (shaded rectangles). Note the out-of-phase cycles recorded from the three VRs, with reduced amplitude and periodicity after the impact
Fig. 8
Impact perturbs the features of electrically induced fictive locomotion. A–D Green box-and-whisker plots describe average values for the main descriptors of fictive locomotor patterns reported from VRrL2 in control and at 15 min, 1 h, and 2 h following the injury. A Cumulative depolarization significantly decreases after impact (*P = 0.002, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 6). B Impact largely reduces the amplitude of oscillations (*P < 0.001, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 6). C Episodes of fictive locomotion are shorter one-hour after the impact (*P = 0.031, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7). D Period of oscillations is significantly smaller 15 min and one-hour post-impact (*P = 0.008, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7). E and F Orange box-and-whisker plots describe values for the main descriptors of fictive locomotor patterns reported from VRrL5 in control and at 15 min, 1 h, and 2 h following the injury. E Periods of fictive locomotion (FL) oscillations 15 min and one hour after injury are significantly shorter than in the control group (*P = 0.002, Friedman test, P = 0.002). F Period CV is higher than in control only at 15-min post-injury (*P = 0.006, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7). G Phase coupling between extensor and flexor commands (homolateral CCF, *P = 0.013, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7) is poorer after the impact. H. Phase coupling between the left and right output (homosegmental CCF, *P = 0.001, repeated measures ANOVA followed by Dunnett multiple comparisons test vs ctrl, n = 7) reduces post injury
In summary, a calibrated impact to the thoracic cord affects the functionality of lumbar locomotor circuits, generating less coordinated locomotor-like oscillations, with shorter and faster cycles of loco
Comments (0)