Cell lines
HUVECs and HDFs were sourced from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. HDFs were incubated in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 40 mM D-glucose, 1% Penicillin-Streptomycin-Amphotericin B Solution (Keygen, China), and 10% fetal bovine serum (FBS). HUVECs were cultured in the medium used for endothelial cells (ECM; ScienCell, USA) containing 40 mM D-glucose and 10% FBS. Standard conditions were provided for all cells (37 °C, 5% CO₂).
Cell transfection
CXXC5 gene-targeting shRNA lentivirus carrying the U6-shRNA-gcGFP-puro plasmid or the control non-specific shRNA lentivirus (GeneChem, Shanghai, China) were transfected into HUVECs and HDFs. After 8 h of transfection, the medium was altered to a fresh medium. 1 µg/ml of puromycin was used for 2 days to construct stable knockout cells and then cultured in DMEM added with puromycin (0.5 µg/ml).
Immunofluorescence assay
Cells were seeded in 24-well plates (3524, Corning, USA) overnight. Phosphate-buffered saline (PBS, 10mM, pH = 7.4, Keygen, China) was rapidly added to rinse the cultured cells twice and later fixed in 4% paraformaldehyde (NCM Biotech, China) at room temperature for 15 min. After rinsing three times with PBS for 3 min, the cells were osmolised with 0.2% Triton X-100 for 10 min (Beyotime, China). After blocking in solution (Beyotime, China) for 1 h under normal temperature, the cells were processed with primary antibodies at 4 °C overnight (Anti-CXXC5, 1:50, Santa Cruz Biotechnology, USA; Phospho-NFκB, 1:800, CST, USA) and secondary antibodies in the dark for 1 h. DAPI (Sigma-Aldrich, USA) was stained for 10 min under normal temperature and rinsed with PBS. Images were captured using a DMi8 Inverted fluorescence microscope (Leica Microsystems, Germany).
Western blot analysis
Cells in 6-well plates (3516, Corning, USA) were collected and lysed by adding RIPA buffer added with 1% protease inhibitors (PMSF) and phosphatase inhibitors (Solarbio, China). Subsequently, the lysate was transferred to an EP tube and subjected to 30 min of centrifugation at 12,000 rpm, 4 °C. The amount of protein in each sample was quantified utilizing the BCA kit (Beyotime, China). Target proteins were isolated by SDS-polyacrylamide gel electrophoresis and then migrated to polyvinylidene difluoride (PVDF) membranes (Millipore, IPFL00010, USA) at a low temperature. Membranes were blotted with primary antibodies overnight at 4 °C on a slow shaker after blocking with 5% bovine serum albumin (BSA) for 1 h at room temperature. Primary antibodies are as follows: Anti-CXXC5 (1:2500, #822172, ZEN-BIOSCIENCE, China), Anti-β-catenin (1:5000, ab32572, Abcam, USA), Anti-COL1a1 (1:1000, #72026, CST, USA), Anti-α-SMA (1:1000, ab124964, Abcam, USA), Anti-CD31 (1:1000, A19014, ABclone, China), Anti-CTBP1 (1:1000, ab129181, Abcam, USA), Anti-VEGFR2 (1:1000, ab134191, Abcam, USA), Anti-VEGFA (1:1000, ab214424, Abcam, USA), Anti-p-IκBα (1:5000, ab133462, Abcam, USA), Anti-IκBα (1:5000, ab32518, Abcam, USA), Anti-p-NFκB (1:1000, #3303, CST, USA), Anti-NFκB (1:1000, #8242, CST, USA), Anti-PCNA (1:5000, ab92552, Abcam, USA), Anti-TCF1/TCF7 (1:1000, #2203, CST, USA), Anti-p-IKKα(Ser176)/IKKβ(Ser177) (1:1000, #2078, CST, USA), Anti-p-IKKα/β(Ser176/180) (1:1000, #2697, CST, USA), Anti-IKKα (1:1000, #61294, CST, USA), Anti-β-actin (1:4000, 81115-1-RR, Proteintech, USA). Membranes were rinsed three times with TBST and reacted with an anti-rabbit IgG Horseradish enzyme labeled secondary antibody (1:30000, ZB-2301, Zhongshan Jinqiao, China) under normal temperature for 1 h. Finally, an enhanced ECL reagent (Vazyme, China) and the Tanon Chemi-Image system were used to image the protein bands.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was purified from cell lysates and cryogenically ground tissue powder using TRIzol reagent (R410, Vazyme, China), followed by cDNA preparation using a reverse transcription kit (R333, Vazyme, China) in accordance with the manufacturer’s instructions. A SYBR qPCR kit (Q712, Vazyme, China) was used to perform quantitative PCR in 96-well plates (402001, NEST, China) under the following settings: pre-denaturation at 95 °C for 30 s, followed by 40 cycles at 95 °C for 10 s, and a reaction temperature of 60 °C for 30 s. The following primer sets were employed: CXXC5, forword 5′-CAAGAAGAAGCGGAAACGCTGC-3′ and reverse 5′-TCTCCAGAGCAGCGGAAGGCTT-3′. GAPDH, forword 5′-ACCACAGTCCATGCCATCAC-3′ and reverse 5′-TCCACCACCCTGTTGCTGTA-3. GAPDH was employed as control for the normalization of target mRNA levels.
Tube formation assay
Matrigel (Corning, USA) was melted at 4°C overnight for gel preparation. A 50 µl of Matrigel volume was added to a 96-well plate (3596, Corning, USA) using pre-cooled pipette tips and maintained for 40 min at 37 °C. HUVECs were seeded at a density of 3 × 104 and cultured with different media. Tube formation was observed 4 h later under a microscope.
Cell counting kit-8 (CCK8) assay
Logarithmic growth stage cells were harvested, resuspended by trypsinization, and plated in 96-well plates (3596, Corning, USA) (2000 cells/well). After adding 10 µl of CCK8 solution (APExBIO, USA) to each well, the cells were incubated for 2 h. Optical density (OD) was read at 450 nm using a microplate reader. The OD value was used to detect the cell proliferation ability of the different groups at different times.
Scratch wound assay
For the scratch wound tests, the cells were first spread in 6-well plates (3516, Corning, USA) until completely covered. After the monolayer was scraped vertically using a 200ul tip, the isolated cells were washed with PBS. The cultured cells were then replenished with basal medium. Cellular wounds were captured on days 0 and 1.
Transwell assay
The target migrating cells were seeded in the upper chamber of the Transwell, and 10% FBS medium was introduced to the bottom compartment (Corning, USA). Subsequently, the cells were placed at 37 °C for 24 h. Cells were maintained in 4% paraformaldehyde for 20 min, immersed in crystal violet (Sigma-Aldrich, USA) for a further 20 min under normal temperature, washed 3 times with PBS, and visualized using an inverted microscope (Olympus, Japan). Three random microscopic areas (magnified 100x) were counted.
mRNA library construction sequencing
TRIzol reagent was added to separate total RNA from HDFs (Invitrogen, Carlsbad, CA, USA), and the resulting material was purified using RNase-free DNase I (Takara, Kusatsu, Japan). The integrity of the RNA was assessed through 1% agarose gel electrophoresis, which was employed to monitor any potential degradation or contamination. RNA quantification was assessed employing an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA), and the quality and intactness of the RNA were evaluated using a NanoDrop spectrophotometer (Thermo Scientific, DE, USA). Sequencing libraries were prepared according to the producer’s guidelines with the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA). To assign sequences to each sample, index codes were then incorporated. In essence, mRNA was isolated from the overall RNA sample by employing poly T-tagged magnetic beads. The purification of the PCR products was conducted on the AMPure XP system, and the qualitative assessment of the library was performed within the Agilent Bioanalyzer 2100 system. The library was prepared on an Illumina Novaseq 6000 platform on behalf of Beijing Allwegene Technology Company Limited (Beijing, China), resulting in paired-end reads of 150 bp.
RNA-seq data analysis
Raws (reads) were initially processed using proprietary Perl scripts to generate clean (FASTQ-formatted) data. This involved the removal of adapter sequences, low-quality sequences, and poly-N sequences from the raw data. The sorting and removal of duplicated reads, along with the merging of the beam alignment results, were conducted using sam-tools v0.1.18 and Picard-tools v1.41. HTSeq v 0.5.4 p3 was employed to enumerate the number of reads that aligned to each gene. Differential expression of both samples was conducted utilizing the DEGseq (2010) R package. The statistically significant enrichment of the differentiation genes in the pathways of the Kyoto Encyclopedia of Genes and Genomes (KEGG) was checked with the KOBAS software. The sequences of the differentially expressed genes (DEGs) were undergone BLAST (blastx) analyses against the genome of a related species, to identify protein-protein interactions that could be taken from the STRING database (http://string-db.org/).
Cytoplasmic and nuclear fractionation
The HDFs were rinsed once with PBS and retrieved using cell scrapers. A nuclear and Cytoplasmic Extraction Kit (R0050, Solarbo, China) was performed to isolate nuclear and cytoplasmic proteins. After protein fractionation, WB was applied to analyze the level of β-catenin protein in the cytoplasm and nuclear. Lamin A + C (1:1000, ab315838, Abcam, USA) and β-actin (1:4000, 81115-1-RR, Proteintech, USA) were assessed as nuclear and cytoplasmic markers, respectively.
Immunoprecipitation (IP)
HDFs and HUVECs were lysed with inhibitor-containing lysate, and the product (500 µg protein) with protein A + G magnetic beads (P2179, Beyotime, China) and appropriate anti-β-catenin antibody (1:30, ab32572, Abcam, USA), anti-TCF1/TCF7 antibody (1:50, #2203, CST, USA), Anti-VEGFR2 (1:20, ab134191, Abcam, USA), Anti-IκBα (1:20, ab32518, Abcam, USA) and Anti-NFκB (1:100, #8242, CST, USA) was rotated overnight at 4 °C in accordance with the manufacturer’s instructions. Membranes were then reacted with secondary antibody (1:2000, A25022, Abbkine, China) under normal temperature for 1 h. After magnetic separation, the products were subjected to WB analysis to evaluate potential protein-protein interactions.
Establishment of diabetic mice model
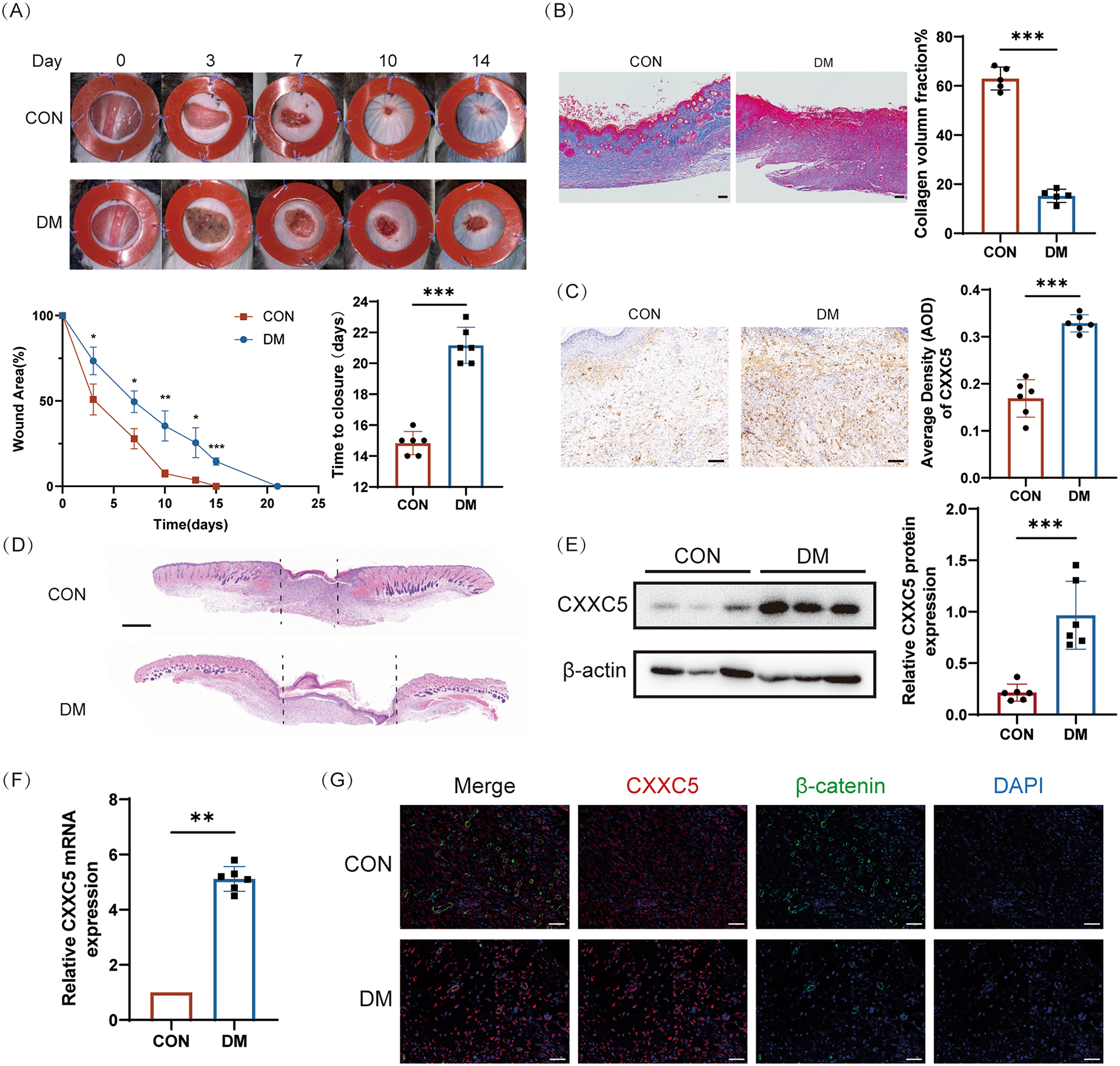
Single Male C57BL/6 mice (6–8 weeks, initial weight about 20 g) were acquired from the Laboratory Animal Center of Nanjing University. In strict accordance with the SPF standard, 6 mice were randomly assigned per cage and adaptively fed for 1 week at the Animal Center of Nanjing Drum Tower Hospital, with 12 h alternating daylight and adequate food and water. The proposed animal experiment has been authorized by the Medical School for Animal Use and Care Committee of Nanjing Drum Tower (Ethics Approval Number: DWSY-22083209) in adherence to ARRIVE guidelines. Mice were randomly (simple randomization) divided into CON, DM, DM + Vehicle, and DM + KY19382 groups (n = 6). After an overnight fast, C57 mice were induced by intraperitoneal injection of streptozotocin (pH = 4.0, dissolved in 0.1 mM citrate buffer, 50 mg/kg) for 5 continuous days. The model was successfully established when the blood glucose level of the mice remained above 16.7 mM for 4 consecutive weeks and a full-thickness incision of 1 cm in diameter was made on the dorsal side of the mice. 2 and 3 mice in the diabetic group died due to hyperglycemia and hypothermia after anesthesia, respectively, while no deaths occurred in CON. A silicone stent with a thickness of 0.6 mm was sutured around the wound to prevent skin contraction, and the dressing was changed daily. DM were intradermally injected with 100 µl KY19382 (HY-131447, MCE, USA) (1mM) [33] on days 0, 2, 4, 6, 8, and 10, while CON were injected with the same volume of Vehicle. The alterations in the wound area of each group were documented photographically and analyzed using the ImageJ software. Wound margin tissue was detected using hematoxylin-eosin (HE) staining, WB, Masson’s trichrome (MT) staining, RT-qPCR, and IF.
Hematoxylin-eosin (HE) and Masson’s trichrome (MT) staining
Once the sections were laid flat, fixed, dehydrated, and paraffin-embedded, a thickness of 5 μm was achieved. The sections were deparaffinized by means of xylene, followed by gradient alcohol hydration, and then stained with HE and MT.
Immunofluorescence (IF) staining
To examine the levels of CXXC5 and β-catenin, the sections above were treated with anti-CXXC5 (1:50, Santa Cruz Biotechnology, USA), anti-β-catenin (1:200, ab32572, Abcam, USA), and anti-Keratin 14 (1:800, 10143-1-AP, Proteintech, USA) at 4 °C overnight, then further stained with fluorescent secondary antibody (1:1000, ab150077, Abcam, USA or 1:1000, ab150116, Abcam, USA) at room temperature for 1 h. After fixing with DAPI (Sigma-Aldrich, USA), the sections were imaged and documented within a TCS SP8 laser confocal microscope (Leica Microsystems, Germany).
Immunohistochemistry (IHC) staining
Paraffin tissue Sect. (5 μm thickness) were deparaffinized, hydrated, antigenically repaired, and exposed to PBS consisting of 0.2% Triton X-100 for 15 min, after which they were cooled to normal temperature. After blockade with 5% BSA, the sections were reacted with anti-CXXC5 (1:50, Santa Cruz Biotechnology, USA), Anti-VEGFA (1:100, ab52917, Abcam, USA), Anti-CD31 (1:600, A19014, ABclone, China) and Anti-p-GSK3β (1:50, ab68476, Abcam, USA), and left at 4 °C overnight. After rewarming, the reaction was repeated with goat anti-rabbit HRP-conjugated secondary antibody (1:20000, ab205718, Abcam, USA) for 1 h at normal temperature. 3, 3-Diaminobenzidine (DAB, 1:20, Sigma, USA) was used for development, and hematoxylin (Solarbio, China) was supplement to stain the nuclear for 1 min. The sections were capped with neutral gum (Solarbio, China). The results were evaluated via a TCS SP8 laser confocal microscope (Leica Microsystems).
Statistical analysis
All data were given as mean ± standard deviation (SD) and statistically analyzed using GraphPad Prism software. Statistical analysis was conducted using Student’s t-test or one-way analysis of variance (ANOVA), and two-way ANOVA. Normality was assessed with the Shapiro–Wilk test. The unpaired Student’s t-test was used for normally distributed data to analyze comparisons between the two groups; otherwise, the Mann–Whitney nonparametric test was used. Multiple pairwise comparisons have been adjusted for significance by the Bonferroni correction. Statistically meaningful results were expressed as (*P < 0.05; **P < 0.01; ***P < 0.001). Results were regarded as non-significant (ns) when the p-value was over 0.05.






Comments (0)