Isolation, culture, and expansion of hUSCs
The isolation and culture of hUSCs were performed according to a previously reported protocol [19]. Briefly, 100 mL of urine was collected from each healthy male participant, with a total of 10 persons aged between 20 and 30 years. Each sample was centrifuged at 400 g for 10 min, and the pellets were resuspended with 10 mL of phosphate buffered saline (PBS) solution, followed by a second centrifugation at 400 g for 10 min. Then, the supernatant was removed as completely as possible, and the cells in the pellets were resuspended with a 1 mL hUSC culture medium containing KnockOut Serum Replacement (KSFM), 22% High Glucose-Dulbecco’s Modified Eagle Medium (H-DMEM), 22% Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12), 10% fetal bovine serum (FBS), 1% insulin-transferrin-selenium (insulin-transferrin-Se), 100 U/mL penicillin and streptomycin (all from Gbico), 10 mol/L cholera toxin and adenine, 2 × 10–9 mol/L 3,3′,5-Triiodo-L-thyronine, hydrocortisone (all from Sigma), and 10 ng/mL epidermal growth factor (EGF) (Peprotech, USA). They were cultured in 12-well plates in a 5% CO2 incubator. The medium was changed daily for 3 days and then it was replaced every 3 days. hUSC colonies typically emerged after 7–10 days of culture, designated as passage 0 (P0). The cells were transferred to a 10 cm dish from a 6-well plate when they reached 80% confluence (P2) for further expansion. Usually, hUSCs from passages 3–5 (P3-P5) were utilized for further experiments.
Preparation of conditioned media and exosomes
hUSCs and DFL (deep fiber layer) cells were cultured in an H-DMEM medium (without serum) for 48 h and then their supernatant was collected and centrifuged at 5000 g for 1 h at 4 ℃ to obtain hUSC-conditioned medium (hUSC-CM) concentrated to 10-fold. For preparing exosomes, the supernatant was centrifuged at 1000 g, 3000 g, and 10,000 g for 10 min at 4 ℃, respectively, followed by a final centrifugation at 100,000 g for 1 h at 4 ℃ using an ultra-high-speed centrifuge (Beckman, USA). The concentrations of hUSC-Exo and DFL-Exo were adjusted to 30 μg/mL, and the exosomes were identified by morphology using electron microscopy, particle size examination with nanoparticle tracking analysis, and specific marker detection with Western blot (WB) analysis.
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was isolated from ovarian granulosa cells (GCs) using the TRIzol reagent (Thermo Fisher, USA), and the RNA was reversely transcribed into cDNA using M-MLV reverse transcriptase (Promega, Shanghai, China). The primers for the target genes are provided in Supplementary Table 1. Polymerase chain reactions (PCR) were conducted in a PCR thermal cycler (Thermo Hybaid, USA). PCR products were subjected to electrophoresis on a 1.0% (mass/volume) agarose gel containing 0.5 μg/mL ethidium bromide for nucleic acid visualization under ultraviolet (UV) light. Human GAPDH served as an internal control. Quantitative PCR was performed using the ABI-ViiA7 PCR machine. The primers for qPCR are listed in Supplementary Table 2. The housekeeping gene, GAPDH, was used to normalize the gene expression.
Flow cytometry analysis
hUSCs were characterized by flow cytometry as follows: the cells were washed and resuspended in PBS (1 × 106cells/mL) and were incubated with antibodies targeting MSC markers (CD90-FITC (561969), CD29-APC (561749), CD73-PE (561,014), and CD105-PE (562380)), hematopoietic cell markers (CD34-PE (560941) and CD45-FITC (560976)), major histocompatibility antigens (HLA-ABC-PE (560168) and HLA-DR-FITC (347363)), and costimulatory molecules (CD80-FITC (557226), CD86-PE (560957), and CD40-FITC (555588)) (all from BD Biosciences) for 30 min. Then, the cells were washed twice with PBS and resuspended in 400 μL of PBS for flow cytometry analysis (Beckman, USA). The results were analyzed using Flowjo version 10.8.1 software. The cells without treatment were used as a negative control, and the region with the highest cell concentration was designated as the P1 area. Subsequently, the position of the cell group in the P1 area was employed as the negative region for the gate, specifically the quadrant 1 lower left (Q1-LL) area depicted in the figure.
Soft agar tumorigenicity test
The bottom layer of the soft agar (0.6%) was prepared in 6-well plates, and the hUSCs were resuspended in 0.3% soft agar at a concentration of 1 × 103cells/well and then plated onto the plates with 0.6% soft agar. They were incubated at 37 °C with 5% CO2 for 30 days. 4T1 cells were used as the control. The colonies were observed and imaged by phase contrast microscopy.
In vivo tumorigenicity test
hUSCs and breast cancer cells (4T1, CBP60352, Nanjing Cobioer Biosciences Co., Ltd.) were suspended in 100 µL of PBS at a concentration of 1 × 106 cells and injected into the breast pads of NOD-SCID mice under anesthetization; these mice were purchased from Changsha SLAC Laboratory Animal Company (Changsha, China, http://www.hnsja.com/) 4T1 cells served as a positive control. Tumor formation was monitored daily for 20 weeks.
Animal models
C57BL/6 female mice, which were 8 weeks old, were purchased from Changsha SLAC Laboratory Animal Company (Changsha, China, http://www.hnsja.com/) and maintained on 12-h light/dark cycles with food and water available ad libitum at the Laboratory Animal Center of Institute of Translational Medicine of Nanchang University. A total of 30 mice were injected intraperitoneally with CTX (50 mg/kg; MCE, USA) for 15 days to establish POF models. All animal procedures described here were reviewed and approved by the Animal Care and Use Committee of Nanchang University.
Lentiviral transduction of hUSCs
hUSCs were seeded in a 6-well plate with 2 mL of complete medium and incubated at 37 °C with 5% CO2. Then, lentiviral transduction was performed once the cell density reached 30–40%. Briefly, the cultured medium of the cells was replaced and cultured with 1 mL of the mixture of complete medium containing polybrene (8 μg/mL) for 24 h. Then, the appropriate volume of virus was used to infect hUSCs. Following the infection, the hUSCs were incubated at 37 ℃ with 5% CO2 overnight and then additionally incubated at 37 ℃ with 5% CO2 overnight after replacing the culture medium with 1 mL of complete medium. Finally, the transfection efficiency was determined by detecting immunofluorescence of green fluorescent protein (GFP) in hUSCs.
Cell transplantation
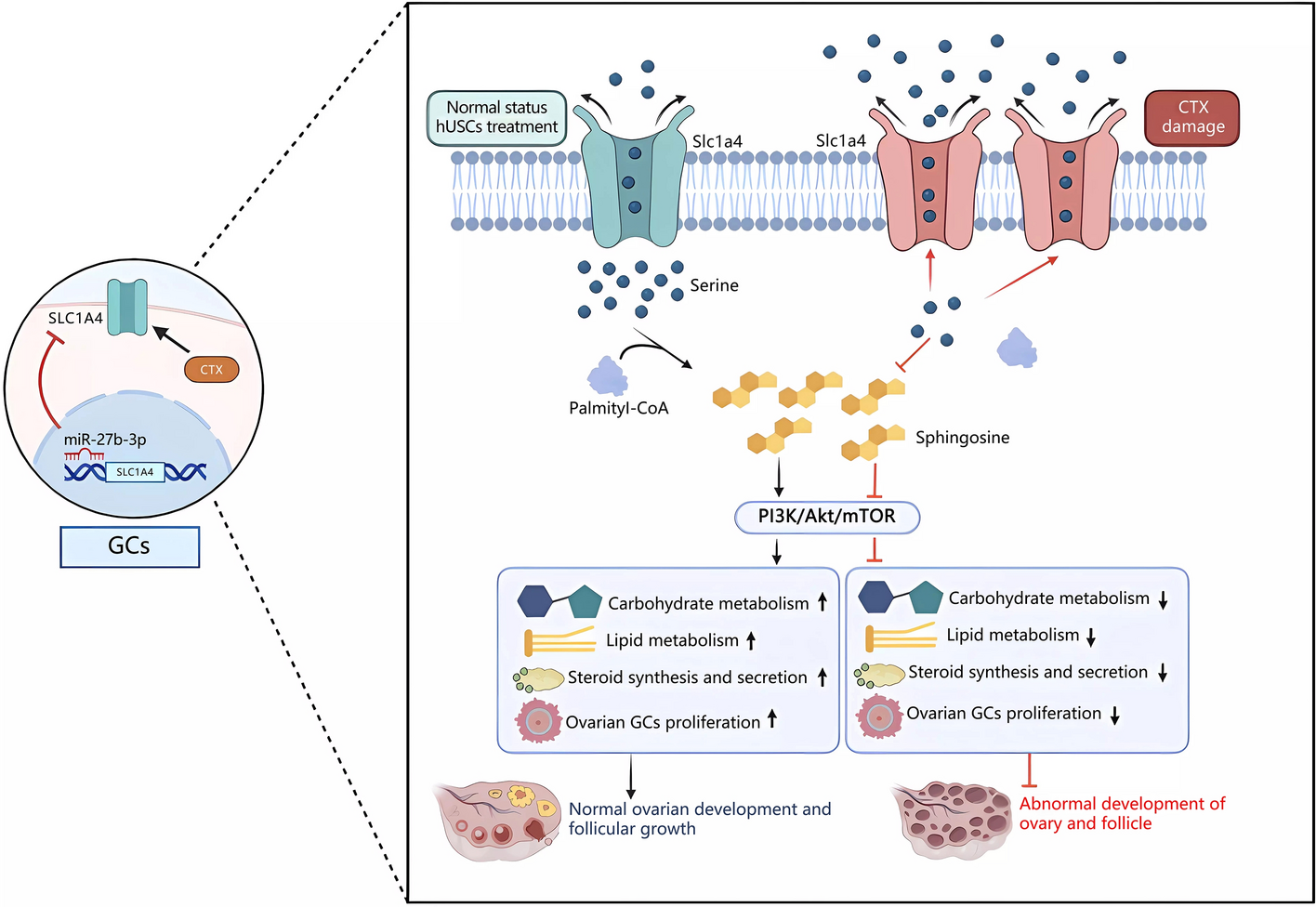
When hUSC confluence reached 85%, the cells were infected with lentivirus labeled with enhanced green fluorescent protein (EGFP) and luciferase at a multiplicity of infection (MOI) rate of 10. Then, 2 days after growth, the cells were examined for EGFP expression by immunofluorescence microscope. In total, 40 mice were randomly divided into four groups with ten mice in each. Mice without the CTX injection served as a control (normal group, n = 10) and the remaining 30 mice were treated with CTX to establish a POF model. The mice treated with CTX were used as the model group (CTX group, n = 10). Subsequently, 1 week later, the modeling mice were injected with hUSCs labeled with GPF (hUSC group, n = 10, 1.5 × 106 hUSCs) or with the concentrated hUSC-conditioned medium (hUSC-CM group, n = 10, 400 μL hUSC-CM) once a week for a total of 3 weeks, respectively. The ovarian tissues and serum of the mice were collected after 1 week for further experiments.
Whole-body fluorescent imaging
After 1, 3, and 7 days of hUSC transplantation, the distribution of the stem cells was examined using whole-body fluorescent imaging system (LB983; Berthold, Germany) in mice. The mice transplanted with hUSCs were euthanized after 1 week, and the liver, heart, spleen, lungs, kidneys, pancreas, and brain were harvested and visualized using the imaging system.
Enzyme-linked immunosorbent (ELISA) assay
Mouse blood samples were kept at room temperature for 4 h and then subsequently left overnight in a 4 °C refrigerator. They were centrifuged at 2000 g for 20 min and the supernatant was collected. Serum estradiol (E2) and follicle stimulating hormone (FSH) levels were measured using an ELISA kit (Cloud Clone Corp, China) according to the manufacturer’s protocol.
Histopathology
Ovarian tissues were collected 1 week after the third cell transplantation, fixed in 4% paraformaldehyde for 24 h, subsequently dehydrated and embedded, and cut into 5 μm thick slices for hematoxylin and eosin staining (H&E staining). The follicles were classified and counted according to the previous study [20]. Follicles in the ovary are divided into four classifications: primordial follicles, primary follicles, secondary follicles, and atretic follicles.
Terminal deoxynucleotidyltransferase-mediated deoxyUTP nick end labeling (TUNEL) assay
The ovaries were fixed in 4% formaldehyde for 24 h and embedded in paraffin. Then, they were cut into 5-μm-thick slices and dewaxed, and apoptosis was detected using a TUNEL kit (Beyotime, China). Five sections of each tissue were examined and analyzed in each experiment.
Western blot analysis
Total proteins were extracted from ovarian tissues using a dedicated total protein kit. Briefly, the ovarian tissues were homogenized in lysis solution (R0010, Solarbio, China) with grinding beads (YA3031, Solarbio, China) and centrifuged at 2000 rpm for 1 min. In addition, the total proteins from GCs and hUSC-Exo were extracted by solely employing a lysis buffer to disrupt both the cells and the exosomes, thoroughly vortaxing the mixture for 30 s and repeating this process five times. The protein concentrations were determined by the bicinchoninic acid method (BCA; PC0020, Solarbio, China). The proteins were run on 10% denaturing SDS-PAGE gels, then transferred to polyvinylidene fluoride (PVDF) membranes (BioRad, USA), which were incubated with primary antibodies at 4 °C overnight. The antibodies used in the study were as follows: anti-GAPDH (1:5000, D4C6R, mouse monoclonal, CST), anti-FSHR (1:1000, CL594-22,665 rabbit monoclonal, protein technology), anti-Bcl-2 (1:1000, D55G8, rabbit monoclonal, CST), anti-P-AKT (1:1000, D9E, rabbit monoclonal, CST), anti-AKT (1:1000, 9272, rabbit monoclonal, CST), anti-P-mTOR (1:1000, D9C2, rabbit monoclonal, CST), anti-mTOR (1:1000, 7C10, rabbit monoclonal, CST), anti-P-PI3K (1:1000, E3U1H, rabbit monoclonal, CST), anti-PI3K (1:1000, 19H8,rabbit monoclonal, CST), anti-CD81 (1:1000, D3N2D, rabbit monoclonal, CST), anti-TSG101 (1:1000, E6V1X rabbit monoclonal, CST), anti-CD63 (1:1000, E1W3T rabbit monoclonal, CST), and anti-CD9 (1:1000, D8O1A rabbit monoclonal, CST). After 16 h, the blots were detected using horseradish peroxidase (HRP)-conjugated goat anti-rabbit or rabbit anti-mouse secondary antibody (Invitrogen, USA) for 1 h at room temperature. The images were quantified using the Super Signal West Pico chemiluminescence detection system.
Immunofluorescence staining
GCs, hUSCs, or ovarian tissue were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100, followed by incubation with the corresponding antibodies at 4 °C overnight. The antibodies used in the study were as follows: anti-FSHR (1:1000, CL594-22665 rabbit monoclonal, protein technology), anti-PCNA (1:200, D3H8P rabbit monoclonal, CST), anti-Oct4 (1:200, D7O5Z mouse monoclonal, CST), anti-SSEA4 (1:300, MC813, mouse monoclonal, CST), anti-Nanog (1:200, D73G4 rabbit monoclonal, CST), and anti-MAB1281 (1:300, mouse monoclonal, 3189191, EMD Millipore, USA). After washing in PBS, the cells were incubated for 1 h at 37 °C with a secondary biotinylated goat anti-rabbit immunoglobulin G (IgG) antibody (dilution 1:300). The cells were then washed in PBS and incubated for 3 min at 37 °C with DAPI dye liquor. The corresponding immunofluorescence staining was recorded with a laser confocal microscope (Olympus CKX41).
Annexin V-propidium iodide (PI) apoptosis assay
For the apoptosis assays, the cells were collected and resuspended in 100 μL annexin V binding solution containing 5 μL annexin V-fluorescein isothiocyanate (FITC) and 5 μL propidium iodide (PI) solution (AD10, Dojindo, Japan). After incubation of 15 min at room temperature, the cells were washed with PBS, centrifuged at 1000 rpm for 5 min, and resuspended in 400 μL annexin V binding buffer. The apoptosis assays were detected and analyzed with BD Jazz.
Live–dead staining
The live–dead staining of cells was performed using the live–dead staining kit (CA1630, Solarbio, China) with a working solution (PBS: diluent: PI: calcein AM = 900:100:1:1), which was added into the medium with the ratio of staining solution (medium = 1:2). The cells were incubated for 15–20 min at 37 °C , being kept away from light, and then detected with a fluorescent microscope.
microRNA (miRNA) sequencing
Total RNA was extracted using the Total RNA Purification Kit (LC Sciences, Houston, USA), according to the manufacturer’s protocol. The total RNA quantity and purity were analyzed using Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with an RNA integrity number (RIN) > 7.0. Approximately 1 μg of the total RNA was used to construct a small RNA library, according to the protocol of the TruSeq Small RNA Sample Prep Kits (Illumina, San Diego, USA). Single-end sequencing (1 × 50 base pairs (bp)) was performed by LC-BIO (Hangzhou, China) using Illumina Hiseq2500.
The differentially expressed miRNAs were identified by volcano plot analysis and heat map analysis. The volcano diagram features the abscissa represented as log2 and the ordinate as –log10. In this diagram, red dots represent significant upregulated miRNAs, blue dots denote significant downregulated miRNAs, and gray dots indicate the miRNAs without differential expression. The heat map utilizes log10 (normalized value) to depict the expression levels of miRNAs with the abscissa representing the samples and the ordinate representing the miRNAs. Various colors on the heat map correspond to different levels of miRNA expression.
Transcriptome sequencing
Total RNAs were used for transcriptome sequencing. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in First Strand Synthesis Reaction Buffer (5X). First strand cDNA was synthesized using a random hexamer primer and M-MuLV reverse transcriptase (RNase H–). Second strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After the adenylation of 3′ends of DNA fragments, adaptors with a hairpin loop structure were ligated to prepare for hybridization to select cDNA fragments preferentially between 370 and 420 bp in length. The library fragments were purified using the APure XP system (Beckman Coulter, Beverly, USA). Then, PCR was performed with Phusion High-Fidelity DNA Polymerase, universal PCR primers, and Index (X) Primer. Finally, PCR products were purified using the AMPure XP system and the library quality was assessed on the Agilent Bioanalyzer 2100 system.
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumia), according to the manufacturer’s instructions. After clustering, the library was sequenced by the Illumina Novaseq platform and 150 bp paired-end reads were generated.
Overexpression of miRNA
Ectopic overexpression of miRNA was achieved by transient transfection of has-miR-27b-3p miRNA mimics (100 nM) (miR10000419-1-5, RIB BIO, China). Transfections were conducted using Lipofectamine® 2000 or Lipofectamine® 3000, following the manufacturer’s instructions. The concentrations of miRNA mimics and transfection reagents were optimized using Alexa Fluor Red Fluorescent Control (RIB BIO, China), along with subsequent miRNA-specific RT-qPCR, utilizing the TaqMan MicroRNA Reverse Transcription Kit from Thermo Fisher Scientific.
Statistical analysis
The results were presented as means ± standard deviations (SD). The unpaired t-test was used for analysis between the two groups. One-way analysis of variance (ANOVA) was used to compare data among three or more groups, as indicated. Differences with a P-value of < 0.05 were considered statistically significant. The software used for data analysis was Prims 10.







Comments (0)