
Remember me




The lifetime risk of heart failure in the United States is approximately 24% and by 2030 it is projected that 8.5 million Americans will have heart failure [1,2,3]. Echocardiography has a preeminent role in the diagnosis, prognostication, surveillance and treatment of heart failure. Insight into cardiac structure and function together with portability and widespread availability has solidified the role of echocardiography as an invaluable decision aid [4, 5]. This review will highlight essential aspects of the echocardiographic assessment that inform the diagnosis and management of cardiomyopathies. The European Society of Cardiology defines a cardiomyopathy as myocardial disease characterized by structurally and functionally abnormal myocardium not caused by coronary artery disease, hypertensive heart disease, valvular heart disease or congenital heart disease. Under this framework, cardiomyopathies are grouped by phenotype as dilated, hypertrophic, restrictive, arrhythmogenic and unclassified [6]. Although coronary artery disease and cancer therapy-related cardiac dysfunction are not classified as cardiomyopathies using this definition, they are important causes of heart failure and will be included in this review.
Hypertrophic CardiomyopathyHypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic heart disease manifested as left ventricular hypertrophy in the absence of causal cardiac, systemic or metabolic disease. Echocardiography is the primary imaging modality in hypertrophic cardiomyopathy. Cardiac MRI (cMRI) adds incremental value in instances of diagnostic uncertainty, poor echocardiographic images and decision making surrounding ICD placement for sudden cardiac death prevention [7]. In addition, cMRI allows superior tissue characterization (useful for exclusion of phenocopies such as infiltrative, metabolic and storage disease) and identification of aneurysms [8].
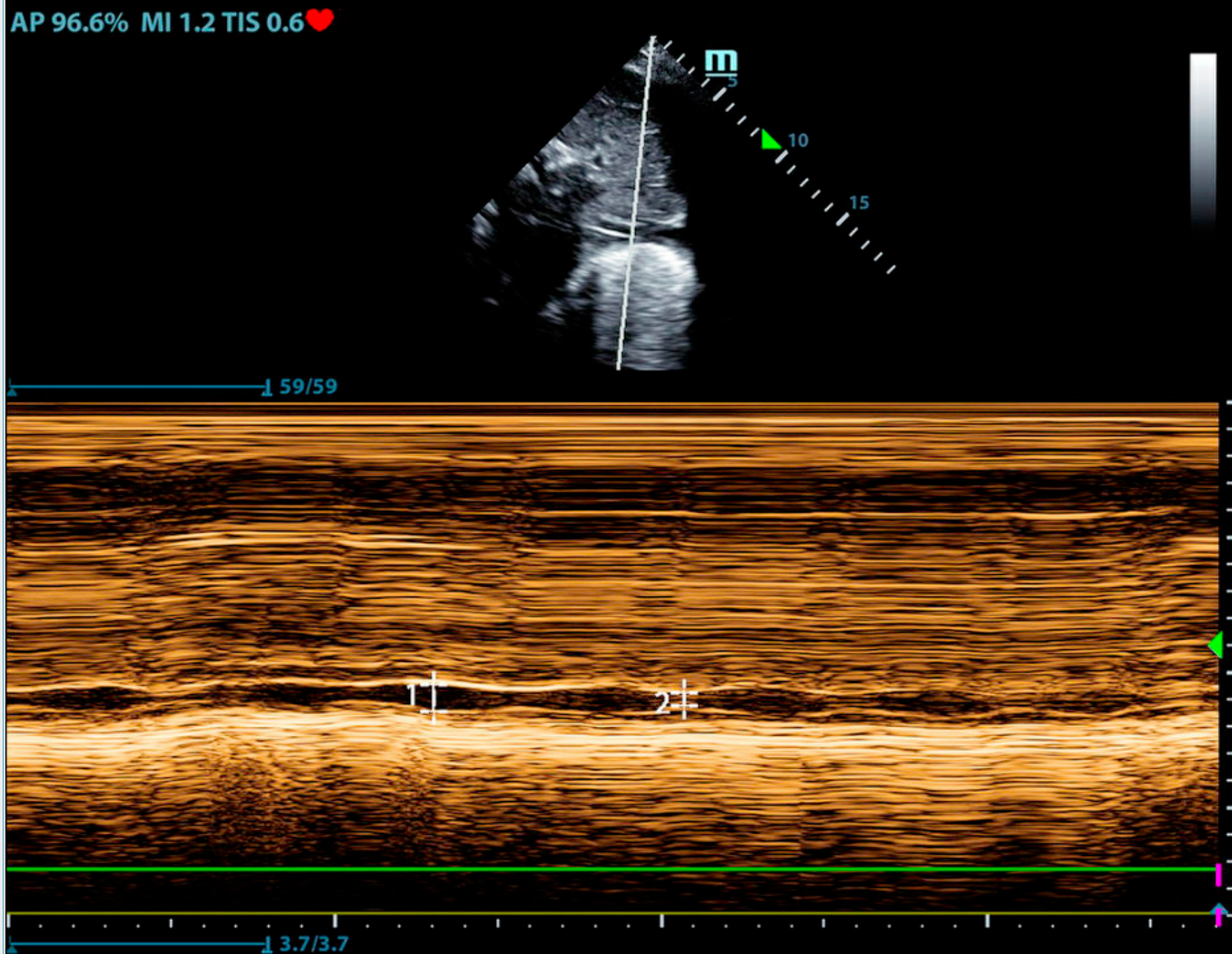
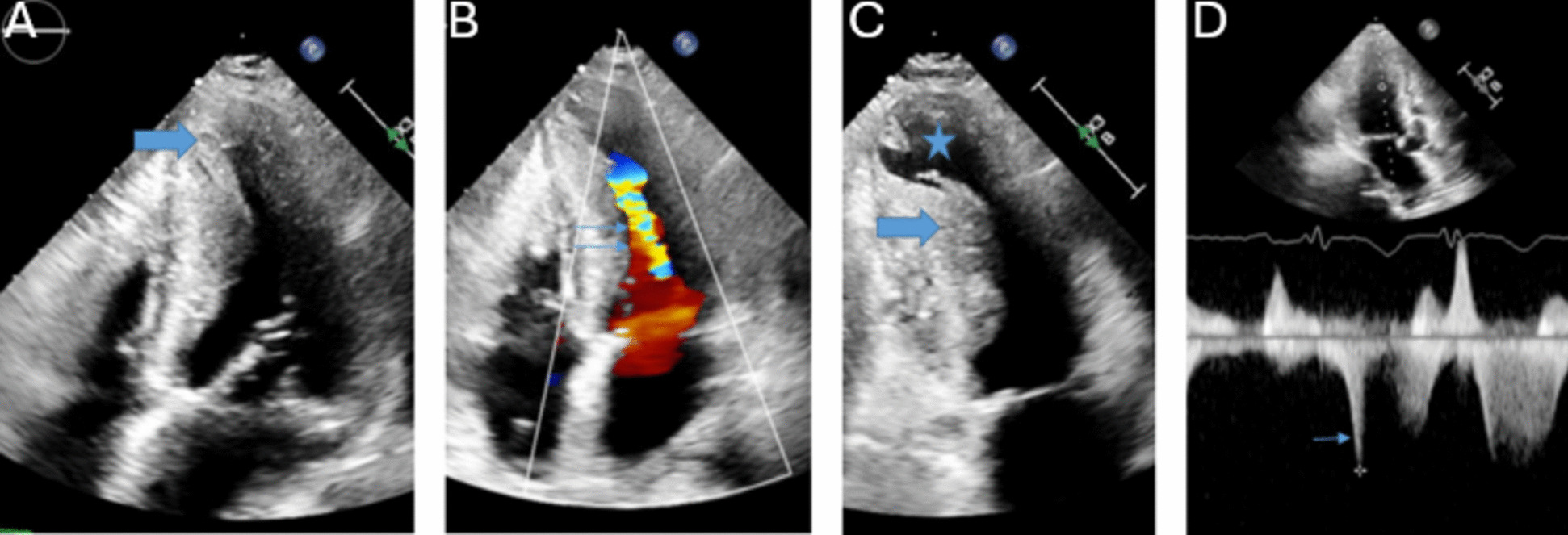
2D/ 3DThe diagnosis of HCM in adult patients requires detection of LV maximal end-diastolic wall thickness ≥ 15 mm at any segment. Left ventricular maximal end-diastolic wall thickness ≥ 13 mm is sufficient for diagnosis in the presence of HCM gene positivity or in patients with family history of HCM [8]. Massive hypertrophy is defined as LV maximal end-diastolic wall thickness ≥ 30 mm and is associated with increased risk of sudden cardiac death [9]. Due to significant heterogeneity in distribution of myocardial thickening, segments should be measured in a combination of short axis and long axis views [10]. Figure 1 depicts a patient with hypertrophic cardiomyopathy. Apical (1 A) and mid-cavitary (1C) hypertrophy are present. Multiple strategies have been proposed to capture maximal wall thickness [9, 11,12,13,14]. The British Society of Echocardiography recommends measurement of septal wall thickness in parasternal long axis and maximal wall thickness on parasternal short axis at basal and mid-LV (at 12, 3, 6 and 9 o’clock positions) and apical level (at 12 and 6 o’clock positions) [15]. The basal anterior septum is the most common site of increased wall thickness [8]. When classic asymmetric hypertrophy is present, a septal to inferolateral wall ratio > 1.3 is often present [16]. Three dimensional echocardiography can increase accuracy of maximal wall thickness measurement and increase ease of apical aneurysm detection [10, 17]. Apical aneurysm detection should prompt consideration for therapeutic anticoagulation given thromboembolism risk [18]. Figure 1 demonstrates an apical aneurysm (1C). Echo contrast functions to improve visualization and is recommended in patients with apical hypertrophic cardiomyopathy [19]. Supportive findings for HCM include systolic anterior motion of the mitral valve, hyperdynamic LV systolic function, myocardial crypts, aneurysms, apically displaced papillary muscles, absent chordae tendinae with anomalous insertion of the papillary muscle into the anterior mitral leaflet, elongated mitral valve leaflets and right ventricular hypertrophy [7].
Fig. 1
This is a patient with HCM. The thick arrows demonstrate apical (A) and mid ventricular (C) hypertrophy. The star denotes an apical aneurysm (C). Color Doppler (double thin arrows) and spectral Doppler (single thin arrow) demonstrate flow acceleration and an intracavitary gradient, respectively (B & D). A 36 mmHg fixed mid-cavitary obstruction was present
DopplerDoppler interrogation is important for the detection of obstruction and mitral regurgitation. Figure 1 demonstrates color Doppler flow acceleration (1B) and spectral Doppler mid-cavitary gradient (1D.) Obstruction can be dynamic and vary with load and contractility. Left ventricular outflow tract (LVOT) obstruction is associated with heart failure and mortality [20]. The predominant mechanisms of LVOT obstruction are septal hypertrophy mediated LVOT narrowing with derangements in blood flow that anteriorly displace the mitral valve leaflets and anatomic variation of the mitral valve and subvalvular apparatus that increases susceptibility of mitral leaflets to abnormal flow vectors [21, 22]. LVOT obstruction is defined as a peak instantaneous gradient ≥ 30mmHg. Gradients ≥ 50mmHg are considered of hemodynamic consequence and in the context of moderate to severe symptoms, invasive septal treatments should be considered [23]. If a gradient of 50mmHg is not detected at rest, with provocation, or with Valsalva, exercise should be performed to unmask obstructive physiology [8]. Mitral regurgitation is mediated by LVOT obstruction (and subsequent systolic anterior motion of the mitral valve) or primary mitral abnormalities. LVOT mediated mitral regurgitation is traditionally eccentric, posteriorly directed, and primarily mid to late systolic due to loss of mitral leaflet coaptation. Non posterior regurgitant jets should prompt work up for intrinsic mitral valve disease [24].
DiastologyDiastolic dysfunction is present in nearly all hypertrophic cardiomyopathy patients. Diastolic dysfunction is mediated by hemodynamic abnormalities, increased LV cavity stiffness and abnormal intracellular calcium uptake [25].
StrainStrain assessment in HCM is notable for reduced global longitudinal strain in the context of preserved ejection fraction. The extent of wall thickness and myocardial abnormalities (fibrosis/ infiltration) are key determinants of global longitudinal and regional strain in patients with LVH [26, 27].
DifferentialIn hypertensive heart disease, LVH is seldom > 18 mm and the pattern of LVH is more likely concentric [28]. In athlete’s heart, diastolic function is normal, LVH is seldom > 12 mm and LV cavity is often dilated > 54 mm (contrasting small cavity of HCM) [29,30,31]. In amyloid, the strain pattern demonstrates apical sparing which distinguishes it from hypertrophic cardiomyopathy [32].
Surveillance & ScreeningThe 2020 AHA/ ACC Guidelines for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy recommends transthoracic echocardiography (TTE) for patients suspected of HCM, and for surveillance of asymptomatic patients every 1 to 2 years. TTE is also recommended for clinical change and 3–6 months after septal ablative therapies. In addition, a TTE or transesophageal echocardiogram (TEE) with intracoronary enhancing contrast injections should be used to assess septal perforators prior to alcohol septal ablation. A TEE is recommended to assess mitral valve structure and function, as well as adequacy of septal myectomy. TTE is recommended as part of initial family screening in first-degree relatives of patients with HCM. Due to the possibility of phenotypic changes, echocardiographic surveillance is recommended in adult genotype positive and phenotype negative first-degree relatives of patients with hypertrophic cardiomyopathy every 3 to 5 years or for change in clinical status [33].
Arrhythmogenic Right Ventricular Cardiomyopathy/ DysplasiaArrhythmogenic right ventricular cardiomyopathy/ dysplasia (ARVC/D) is a largely genetically mediated disease characterized by the fibrofatty replacement of the myocardium of the right ventricle, left ventricle, or both ventricles [23, 34]. Cardiac MRI is recommended in all patients in whom there is clinical suspicion for ARVC/D [35].
2D/ 3DA comprehensive echocardiographic assessment should include assessment of the parasternal long axis (PLAX), right ventricular outflow tract (RVOT), PLAX RVOT index, parasternal short axis (PSAX), RVOT, PSAX RVOT index, RV basal diameter, and RV fractional area change [36]. Advanced disease may have diffuse biventricular involvement [37]. The Padua criteria defines a right ventricular and left ventricular category (arrhythmogenic left ventricular cardiomyopathy). The right ventricular major criteria are regional RV akinesia, dyskinesia, or bulging plus global RV dilatation or global RV systolic dysfunction. Minor criteria are regional RV akinesia, dyskinesia or RV free wall aneurysm. Left ventricular minor criteria are global LV systolic dysfunction (LVEF or GLS) with or without LV dilatation. Additional minor criteria are LV hypokinesis of LV free wall, septum or both [38]. 3D echocardiographic assessment of right ventricular volumes and RV ejection fraction should be considered when proper software and expertise are present [36]. 3D right ventricular ejection fraction ≤ 40% is considered abnormal [36].
StrainLV GLS and RV longitudinal strain of lateral RV free wall should be performed. Longitudinal strain of the lateral RV free wall worse than − 23 is considered abnormal. LV GLS strain worse than − 18 is considered abnormal [36]. Right ventricular deformation imaging performed in apical 4-chamber view adds insight to screening of 1st degree relatives. Abnormal right ventricular speckle tracking is associated with disease progression [39].
DopplerComprehensive assessment should include tricuspid annular plane systolic excursion (TAPSE) and assessment of tricuspid regurgitation [36].
Screening & SurveillanceThere is considerable variability in disease progression [40]. Patients with ARVC/D should be monitored with serial cardiac magnetic resonance imaging (cMRI) or serial echocardiography. Serial echocardiography should be performed every 1 to 3 years based on age and clinical and/ or genetic features [41]. Echocardiographic surveillance of 1st degree relatives of ARVC/D patients is essential. Approximately a third of 1st degree relatives will develop ARVC/D. 1st degree relatives should be screened every two to three years and annually for those engaged in competitive or endurance sports [42].
Restrictive CardiomyopathyRestrictive cardiomyopathy (RCM) is caused by a diverse group of myocardial disorders united by a common phenotype. The restrictive phenotype is characterized by left ventricular noncompliance, impaired diastolic filling and elevated filling pressures. RCMs are classified as infiltrative disease, storage disorders, interstitial fibrosis/ intrinsic myocyte dysfunction and endomyocardial diseases [43].
2D/ 3DThe hallmark of the 2D assessment is biatrial dilatation (not attributed to valvular heart disease or atrial fibrillation) and non-dilated ventricles. Left ventricular systolic function is usually normal or near normal and wall thickness can vary [44]. However, reduced left ventricular systolic function can occur with advanced disease [45].
DopplerSupportive features include congestion of the inferior vena cava and hepatic veins. In addition, there may be hepatic vein diastolic inspiratory reversal due to the inability of stiff right ventricle to handle increased venous return with inspiration. In addition, increased stiffness of the left heart can lead to pulmonary hypertension [43, 45].
DiastologyDoppler interrogation of the mitral valve inflow demonstrates a restrictive filling pattern. The E wave is elevated due to increased early diastolic filling mediated by elevated left atrial pressure. The A wave is reduced due to high ventricular diastolic pressure. The mitral deceleration time and the isovolumetric relaxation time are reduced. Tissue Doppler velocities are reduced. High atrial filling pressures and decreased ventricular compliance result in decreased systolic to diastolic pulmonary venous flow ratio [46, 47].
DifferentialConstrictive pericarditis is a potentially curable form of heart failure due to the hemodynamic changes of a stiff pericardium. The abnormal pericardium impairs filling and causes biventricular diastolic dysfunction and abnormal filling pressures. Both RCM and constrictive pericarditis have elevated filling pressures and diastolic dysfunction. Echocardiography can assist in distinguishing between restrictive cardiomyopathy and constrictive pericarditis. There are several hallmark echocardiographic findings of constrictive pericarditis. They are respiratory-related ventricular septal shift (septal bounce), accentuated respiratory variability in mitral inflow E velocity, preserved medial mitral annular e’ velocity, annulus reversus, and hepatic vein expiratory diastolic flow reversal [45, 48, 49].
Cardiac AmyloidosisThe majority of cases of cardiac amyloidosis can be attributed to amyloid light chains or transthyretin protein [50, 51].
2D/ 3D/ DopplerMyocardial infiltration results in ventricular thickening. LV wall thickness > 1.2cm without alternative cause should raise suspicion for cardiac amyloidosis [52]. The pattern of pseudohypertrophy is often asymmetric in ATTR amyloidosis and symmetric in AL amyloidosis [43]. Right ventricular involvement is associated with worse prognosis. Supportive findings for cardiac amyloidosis include the presence of thickened valves (> 0.5 cm), thickened interatrial septum, sparkling myocardium appearance and the presence of a pericardial or pleural effusion [44, 52]. Tissue Doppler velocities (s', e' and a’) all < 5cm is suggestive of cardiac amyloidosis (5-5-5 sign.) [52]. In ATTR amyloidosis worsening MR and TR are independently associated with mortality [53].
StrainGlobal longitudinal strain is decreased, or less negative in cardiac amyloidosis. Infiltration (and impairment) is greatest at the bases and decreases toward the apex (basal-apex gradient). This results in the pathognomonic cherry on top sprain pattern with apical sparing [43]. Strain has prognostic implications. In patients with AL amyloidosis peak longitudinal systolic basal anteroseptal strain less negative than or equal to -7.5% was described as high risk by Bellavia et al. [54].
DifferentialIn contrast to HCM or hypertensive heart disease, myocardial infiltration often results in thickening of the right ventricular free wall and atrial septum [55].
Screening & SurveillanceSurveillance imaging is indicated, frequency of which should be determined by amyloid type and clinical picture [56]. Individuals suspected of having cardiac amyloid, relatives of individuals with TTR variants and TTR genotype positive/ phenotype negative individuals should be considered for screening with TTE, bone scintigraphy imaging and cardiac MRI. Screening for relatives should be initiated within 10 years of the predicted phenotype onset associated with the TTR mutation [56].
Cardiac SarcoidosisCardiac Sarcoidosis is an inflammatory disorder of the myocardium caused by granulomatous infiltration.
2D/ 3DCardiac sarcoidosis can present as a dilated or restrictive cardiomyopathy. Echocardiographic features of cardiac sarcoidosis include regional wall motion abnormalities in a non-coronary distribution, regional wall aneurysms (predilection for inferolateral wall), pericardial effusion and basal septal thinning/ scarring (IVS thickness ≤ 4 mm at a distance of 10 mm below the aortic annulus) [57, 58]. Figure 2 demonstrates a mid inferior wall aneurysm. Echo contrast can assist with detection of aneurysms. Edema or granulomatous deposition can cause left ventricular wall thickening, and or increased echogenicity of the left ventricular wall [57]. Granulomatous deposition is most common in the right ventricle (46%) basal ventricular septum (73%), lateral free wall (96%) [59]. RV dilatation can occur, but is likely secondary to pulmonary hypertension from pulmonary sarcoidosis. RV aneurysms can be present [44, 60]. However, the presence of a normal echocardiogram does not exclude cardiac sarcoidosis given limited sensitivity and specificity. Thus, assessment should include cardiac MRI and/ or FDG-PET depending upon clinical suspicion [60, 61]. In sarcoidosis, valvular heart disease (tricuspid regurgitation or pulmonary regurgitation) is commonly secondary to pulmonary hypertension. Valve disease from infiltration of papillary muscles is uncommon [57, 62].
Fig. 2
Mid inferior wall aneurysm (arrow) in a patient with cardiac sarcoidosis
DopplerPulmonary hypertension mediated by pulmonary involvement or LV dysfunction occurs in a significant amount of patients with cardiac sarcoidosis [63]. In the context of poor tricuspid Doppler signal, agitated saline can help delineate Doppler envelope for estimation of pulmonary hypertension [57]. It is estimated that 1 in 8 aortic stenosis patients referred for transcatheter aortic valve replacement have bone scintigraphy evidence of amyloidosis. Aortic stenosis assessment should be performed in all patient with cardiac amyloidosis [64].
StrainStrain in cardiac sarcoidosis is reduced (less negative) with an abnormal septal strain pattern. Strain can improve sensitivity in early diagnosis/ detection of preclinical disease. Strain analysis also has prognostic indications [16, 65, 66].
Screening & SurveillanceDifferent screening protocols are recommended based on the clinical picture. Patients with cardiac alarm features (unexplained advanced AV block/ sustained VT/ unexplained LV impairment or incidental CMR pattern suggestive of CS) should begin imaging screening for cardiac sarcoidosis with cardiac MRI +/-FDG PET [67]. Patients with extracardiac sarcoidosis with cardiac symptoms, but without alarm features, should begin cardiac imaging evaluation with TTE with further testing determined by clinical picture [60, 67,
Comments (0)