Neurologists, particularly movement disorders specialists, are trained to classify and discriminate disease entities based on the finest details of clinical phenomenology. Beyond the mere exercise of diagnostic skills, the definition of an accurate movement disorder phenotype is the essential starting point for localizing the site of brain damage and guiding appropriate diagnostic workup and symptomatic therapy. With the genetic revolution, the recognition of clear-cut movement disorder syndromes has also been instrumental in the identification of several disease genes in a “forward genetics” approach using linkage analysis. Paradigmatic examples are the discovery of SGCE as main gene for the syndrome “myoclonus-dystonia” [1] and RFC1 as the gene associated with the triad cerebellar ataxia, sensory neuropathy and vestibular areflexia (CANVAS) [2].
In an opposite “reverse genetics” approach, an ever-growing list of monogenic etiologies for early-onset, clinically less well-defined phenotypes, commonly referred to as “neurodevelopmental disorders” (NDDs) has been unveiled [3]. The term NDDs was introduced in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) as an overarching category for a group of conditions with onset in the developmental period that result in functional impairment in multiple domains [4]. NDDs include intellectual disability, communication disorders, autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), specific learning disorder, and motor disorders (including developmental coordination disorder, stereotypic movement disorder, and tic disorders) [4]. The definition of NDDs also extends to other conditions outside the domain of DSM-5, such as cerebral palsy (CP) and epileptic encephalopathies [5]. The frequency of comorbidity among the NDDs is higher than that expected by chance [6] and provides the rationale for lumping them in a clinical continuum [7]. Due to the extremely heterogeneous and mostly non-specific clinical pictures, NDDs are usually tackled with a “genotype-first” approach using chromosomal microarrays and unbiased exome sequencing [3]. The latter tool has greatly accelerated gene discovery in NDDs [8] and highlighted the molecular overlap with other seemingly unrelated phenotypes, such as adult-onset movement disorders [9,10,11,12,13,14,15]. As a result, our perspective is gradually changing. From a dichotomous paradigm distinguishing neurodevelopmental dysfunction and neurodegeneration, cumulative evidence outlines a nuanced clinical spectrum due to genetically determined developmental brain dysfunction [6], whose modulating factors remain elusive.
The present review focuses on the shared molecular landscape of movement disorders and NDDs. We begin our discussion with the genetic discoveries in CP, the epitome of disease of the movement and the developing brain. We then review selected biological pathways that emerged as common culprits of neurological dysfunction both in early development and later in life. Finally, we discuss the clinical implications of the increasing NDDs-gene etiologies in movement disorders.
The Cerebral Palsy Paradigm
Cerebral palsy (CP) is a clinical diagnosis describing neurodevelopmental phenotypes that primarily affect movement and posture [16]. CP is attributed to nonprogressive disturbances occurring early in the fetal or infant brain [16]. Birth asphyxia secondary to intrapartum complications has long been considered its leading cause [17,18,19]. Large-scale genetic studies using chromosomal microarray analysis and subsequently exome or genome sequencing have challenged this dogma, demonstrating a genetic etiology in 31.1% of cases on average [20]. The diagnostic yield of exome sequencing may approximately double if CP cases without hints of perinatal brain injury according to clinical history and/or brain MRI are selected [21]. When the broad clinical umbrella of CP is re-evaluated based on the clinics, the presence of a hyperkinetic movement disorder phenotype (dystonic and/or dyskinetic) is another predictive factor for a monogenic etiology [22]. Similar to other NDDs [23,24,25,26], the rate of de novo variants in CP is high [21] and may explain the relatively constant frequency of these disorders associated with reduced fitness despite the improvement of perinatal care in developed countries [27].
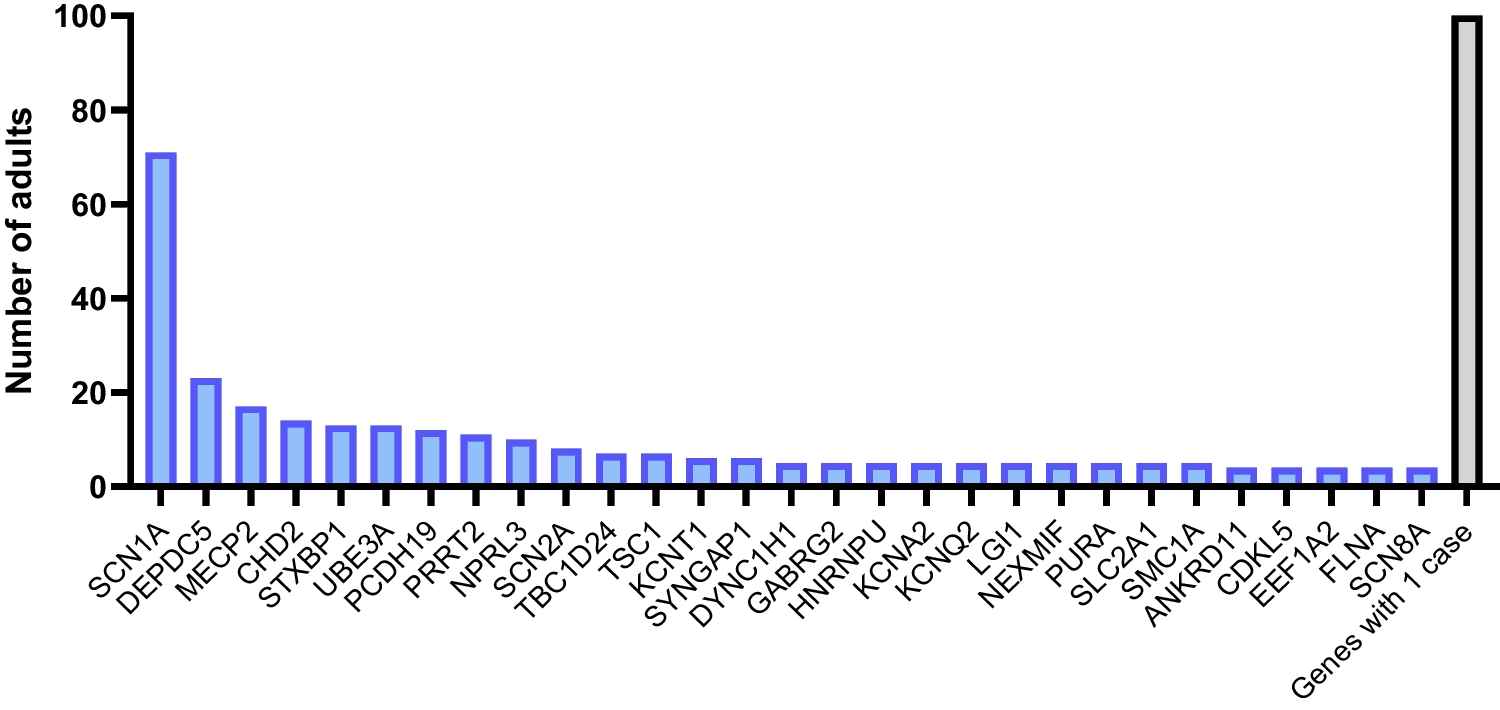
Looking at the molecular pathways involved, the most common monogenic etiologies associated with CP cluster in a few complex processes with a key role in neurodevelopment, such as transcriptional regulation (CTNNB1, FOXG1, MECP2), neuritogenesis (ATL1, KIF1A, SPAST, TUBA1A, TUBB4A), and synaptic transmission (CACNA1A, GNAO1, KCNQ2, SCN1A) [20]. Notably, several of these genes have been previously implicated in classic adult-onset movement disorders, such as autosomal dominant TUBB4A-related dystonia [28, 29], spastic paraplegia type 4 (SPAST) [30], or inherited cerebellar ataxia phenotypes (CACNA1A) [31, 32]. In the following sections, we will focus on these three overarching biological processes and their involvement in both NDDs and specific movement disorder phenotypes.
Transcriptional Dysregulation as Driver of Neurodevelopmental Brain Dysfunction
Complex processes underlying neurodevelopment and neural function throughout life depend on the coordinated expression of myriads of genes in specific cells at the appropriate time [33, 34]. Beyond the large number of players at a purely genetic level, the ultimate phenotypic complexity underlying neural function is determined by a multifaceted regulation of gene expression. Thus, it is not surprising that an increasing number of genes with DNA-, RNA-, and histone-binding functions are emerging in the landscape of NDDs [35].
Sequential expression of different transcription factors in specific time windows drives the differentiation of neural precursors [33]. For example, NKX2-1 expression in neural progenitors is required for GABAergic interneuron commitment [36] and basal ganglia development [37]. NKX2-1 (Mendelian Inheritance in Man (MIM) *600635) is a well-established human disease gene, initially associated with thyroid and lung developmental defects and later, also with neurological symptoms. One group identified five index patients with additional neurological features such as choreoathetosis, muscular hypotonia, ataxia, and developmental delay in the screening of patients with congenital hypothyroidism, which did not respond to substitution with L-thyroxine, prompting a search for a differential diagnosis [38]. In the same year, another independent group published the association of NKX2-1 variants with the well-known clinical entity “benign hereditary chorea”, a childhood-onset form of chorea not associated with intellectual decline (see also Table 1) [39].
Beyond direct gene activation and repression, chromatin modification offers another level of control on a large scale. DNA-binding proteins that recruit chromatin- and RNA-modifying factors, such as those of the CHD family, have an established role in NDDs [23, 40, 41] and an emerging role in movement disorders [11, 42]. Perhaps the most interesting converging biological pathway in NDDs and movement disorders is DNA methylation, a key regulatory process affecting both ends of the life course [34]. In the zygote, a wave of demethylation occurs prior to methylation imprinting [43]. Alterations in this dynamic process have been implicated in NDDs such as Rett, Prader-Willi and Angelman syndromes [34]. On the other hand, the extent of methylation later in life has been shown to be consistent with the concept of an “epigenetic clock” as a strong predictor of life expectancy [44]. Methylation and demethylation of lysine residues on histone tails is a key dynamic chromatin modification that is mediated by specific methyltransferases (KMTs) and demethylases (KDMs) [45]. Twenty-seven KMT- and 24 KDM-encoding genes are known, and to date, 22 have been associated with NDDs [35]. KMT2B (MIM *606834) encodes histone lysine N-methyltransferase 2B, an epigenetic writer that, like other KMT2 enzymes, modulates transcriptional regulation by methylating a specific lysine residue (K4) of the histone 3 (H3) protein [46]. H3K4 methylation by KMT2B is associated with active transcription and plays an essential role in the normal development and maturation of brain circuits involved in motor control [46,47,48]. The first association between KMT2B and human disease was described in patients with childhood-onset isolated dystonia carrying heterozygous loss-of-function variants [49, 50]. Cumulative reports gradually revealed a much broader phenotype in which developmental features may represent the first or predominant manifestation (see also Table 1) [15, 45], as opposed to adult-onset incompletely penetrant dystonia at the other end of the clinical spectrum [51]. Recently, a unique DNA methylation pattern at CpG sites in peripheral blood from KMT2B patients was described, as the so-called epi-signature [52]. This unique biomarker corroborated some of the genotype-phenotype correlations observed in KMT2B-related disease. For instance, the KMT2B missense variant p.Ala1541Val associated with adult-onset dystonia [51] caused more subtle methylation changes compared to truncating variants seen in early-onset, developmental cases [52]. The importance of proper H3K4 methylation dosage in normal development is further highlighted by the involvement of at least six KMT2 genes in human disease, despite their seemingly redundant enzymatic function [45, 53, 54]. Elucidating the relationship between dysregulated KMT2 function and neurological disease is of particular interest for the development of therapeutic strategies. Indeed, methylation is a dynamic and potentially reversible or inducible process, as suggested by the striking therapeutic effect of deep brain stimulation in the setting of certain KMT2B variants [15].
Defective Neuritogenesis in Developmental Motor Disorders
Neuritogenesis is a crucial step in neurodevelopment [55]. Early-stage neurons appear as round bodies, in which the growth of actin-rich filopodia and lamellipodia marks the step to the acquisition of cellular polarity [56]. Stabilization by microtubules leads to the development of neurites, which then differentiate into axons and dendrites as the cell acquire their mature neuronal morphology [55]. Proper neurite formation is essential for establishing neuronal morphology such as arborization and synapse formation, which in turn influences connectivity in the brain. The same guidance molecules play an important role in directing axonal growth and influencing synaptic plasticity during development and later in life [57]. As such, variants in axon guidance genes have been implicated in both developmental conditions and neurodegenerative diseases. Cumulative evidence pointed out inappropriate connectivity due to abnormal neuronal density, dendritic arborization and/or cortical layering as one of the causes of ASD [58, 59]. Disturbances of neuritogenesis is also a recurring leitmotif in motor disorders with predominant pyramidal tract dysfunction. Variants in proteins involved in microtubule dynamics (SPAST), axonal maintenance (ATL1) and transport (KIF1A) are among the most common genetic etiologies both of cerebral palsy mimicries [20] and of hereditary spastic paraplegia (HSP) [60]. HSPs are progressive, neurodegenerative disorders with later, often adult, onset in many cases [61]. Spasticity in the lower limbs is the most prominent clinical sign, which can occur isolated or in combination with several other neurological features [61]. Notably, early onset with protracted clinical stability has previously been identified as an endophenotype in a subset of patients in HSP families, resembling the non-progressive course of CP [61].
The selective susceptibility of motoneurons to defect of neuritogenesis is plainly explained by their characteristic morphology: extremely long axons with extensive terminal branching. This pose exceptional challenges for the targeted delivery of presynaptic components from the soma, where they are mostly synthetized, as well as for the removal of defective organelles which must be retrogradely transported. Motor proteins such as kinesins and dyneins, along with several adaptors and scaffolding elements, are in charge of the bidirectional transport of synaptic cargos to ensure precise assembly, maintenance, and remodeling of synapses [62]. At least 23 genes coding for such cargo machinery have been associated with NDDs [62]. A particularly broad phenotypic spectrum is associated with variants in the KIF1A (MIM *601255) [63], a kinesin responsible for the anterograde transport of synaptic vesicle precursors along axonal and dendritic microtubules (see also Table 1) [63]. More than 100 disease-associated KIF1A variants have been described in the literature [64]. The broad spectrum of clinical symptoms encompasses both neurodevelopmental and neurodegenerative categories such as developmental delay, intellectual/learning disability, autism, epilepsy, microcephaly, spastic CP, HSP, peripheral neuropathy, optic nerve atrophy, and cerebellar atrophy [64,65,66,67,68]. Considering that anterograde transport of presynaptic components is required for both development of the brain and maintenance of axons functionality through life, this variability is not surprising. KIF1A variants can be dominantly and recessively inherited, as in HSP families, or appear de novo in the most severe phenotypes [64, 69]. Some further genotype-phenotype correlations are known. For instance, patients carrying missense variants at the position 13 (such as R13H and R13C) are at a high risk of ASD [65, 67].
Synaptic Dysfunction in Focus: the Example of CACNA1A Disease Spectrum
Proper neural morphogenesis and branching is instrumental to the development of brain connectivity. The human central nervous system contains ∼1015 synapses between ∼1012 neurons, building a hyper-wired interconnectome [70]. Intense synaptogenesis occurs during embryonic and early postnatal stages, persists throughout adolescence and up to the third decade [71]. Not surprisingly, disruption of synaptic transmission and plasticity leads to a wide range of NDDs.






Comments (0)