Cell line
Murine astrocytoma cell line, ALTS1C1 (T8239, Applied Biological Materials, Richmond, Canada, or BCRC60582, Hsinchu, Taiwan), was previously established in our lab [16]. GL261, a murine glioma cell line, was kindly provided by Dr. Newcomb’s lab at New York University Medical Center. ALTS1C1-GFP cell line was created by lentiviral infection of GFP-expressing vector into the ALTS1C1 cells. ALTS1C1, ALTS1C1-GFP, and GL261 were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, 12100-046, NY, USA) with 10% Fetal bovine serum (FBS; Gibco, 16000-044) and 1% Penicillin–Streptomycin (Gibco, 15140-122). Cells were passaged every 2–3 days and maintained in a humidified incubator at 37 °C with 5% CO2. For cell passaging, cells were first washed with 1X PBS (Biological Industries, 02-023-5A, Israel) and dissociated by 0.5% Trypsin–EDTA (Gibco, 15400-054) at 37 °C for 5 min.
Animal
Six to eight-week-old C57BL/6J male mice were purchased from the National Laboratory Animal Center of Taiwan. All experimental procedures were approved by the Institutional Animals Care and Use Committee (IACUC approval No. 107042) of National Tsing Hua University, Taiwan. All surgeries were performed under anesthesia conditions, and efforts were made to minimize the mice’s suffering. 75% Zoletil 50 (50 mg/mL) (Virbac, France) and 25% Rompun 20 (Bayer, Korea) mixture was used for mouse surgery. Mice were anesthetized by intraperitoneal injection of 2 μL/g Zoletil–Rompun mixture.
Intracranial injection
Tumor cells were counted and resuspended in DMEM. The cell suspension was kept on ice during the surgery. Animals were anesthetized with a Zoletil–Rompun mixture. The fur of the skull was removed, and the animals were fixed on the stereotactic frame (David Kopf Instrument). A midline scalp incision was created, and a burr hole was made at 1 mm posterior and 2 mm lateral (right) from the bregma by a rotary drill. 2 μL tumor cells (1 × 105 cells/2 μL) were injected into the hole at 2.5 mm depth. The cell suspension was delivered through a microinjection needle at 1 μL/min for 2 min. After injection, the needle was left in the brain for 2 min and then slowly removed from the brain. The burr hole was covered with bone wax (ETHICON, W810, Belgium), and the incision was sutured (UNIK SURGICAL SUTURES MFG, ST174, 75 cm, 30″, New Taipei City, Taiwan). The mice were killed on day 18 after tumor injection.
Tissue collection for histological analysis
When killed, mice were anesthetized with a Zoletil–Rompun solution. Then, intracardiac perfusion was performed with cold PBS (8 mL, 2 min) followed by cold 4% paraformaldehyde (PFA) (Sigma, 16,005, Germany) (8 mL, 10 min) to fix the tissue. The brain was removed and soaked in 10 mL PFA at 4 °C 18 h for post-fixation. After post-fixation, the brain tissue was transferred to 25 mL 10% sucrose (Sigma, S0389, USA) at 4 °C 2 h and then to 25 mL 20% sucrose at 4 °C 18 h to dehydrate the sample. After dehydration, the tissue was transferred to 5 mL Optimal cutting compound (OCT; Leica, 14,020,108,926, United Kingdom) at 4 °C 2 h to acclimate the OCT compound. After acclimation, the tissue was transferred to a cryomold filled with OCT and left to balance at room temperature (RT) for 30 min. The cryomold was then covered with lab tape to avoid isopentane contamination. The tissue was frozen by isopentane (ACROS Organics, 126,470,010, USA), cooled by liquid nitrogen for 3 min, and then stored at − 80 °C. Frozen tissue was first thawed at − 20 °C 1 h before cryosection. The tissue was sectioned into 10 μm slices by the cryostat (Leica, CM1850, IL, USA) at − 20 °C and adhered to the saline slide (MUTO PURE CHEMICALS, 511,614, Japan). The slide was then stored at − 80 °C.
Histological analysis—Filipin III staining
The slide was fixed with 1.2% PFA at RT for 10 min and washed 3 times with PBS for 2 min. The tissue was then surrounded by a hydrophobic barrier pen (VECTOR LABORATORY, H-4000, CA, USA). The slide was incubated with 100 µL Filipin III staining solution (250 μg/mL Filipin III in PBS) (Filipin III: Sigma, SAE0087, USA) at RT for 1.5 h and washed 3 times with PBS for 2 min. 100 µL Propidium iodide solution (1 μg/mL Propidium iodide solution in PBS) (Propidium iodide: Sigma, P4170, USA) was added to label the nuclei at RT for 5 min. Following Propidium iodide staining, the slide was washed 3 times with PBS for 2 min and let dry. Finally, 10 μL mounting medium (VECTOR LABORATORY, H-1000-10) was dropped on each tissue slice, and the slide was covered with a coverslip. The image was viewed by a fluorescence microscope (ZEISS, Axioskop 40 CFL, Gottingen, Germany) and captured by the camera (ZEISS, Axiocam 503 color).
Histological analysis—hematoxylin and eosin (H&E) staining
The slide was fixed with 95% methanol (Merck, 33213-2.5L, Germany) at − 20 °C for 10 min and washed twice with PBS for 5 min. The slide was dipped in Hematoxylin solution (Surgipath, 110,619, Leica Biosystems, IL, USA) for 1 min at RT and rinsed with water. The slide was then dipped in 0.25% ammonium (J.T.Baker, 9721-03, USA) for 10 s and rinsed with water. After air-drying, the tissue was dipped in Eosin solution (Surgipath, 82,219, Leica Biosystems) for 20 s at RT and rinsed with water. The slide was dipped twice in 95% ethanol (Merck, 32221-2.5L, Germany) and air dried. Finally, 10 μL mounting medium (VECTOR LABORATORY, H-5000) was dropped on each tissue slice, and the slide was covered with a coverslip. The image was viewed by a microscope (ZEISS, Axioskop 40 CFL, Gottingen, Germany) and captured by the camera (ZEISS, Axiocam 503 color).
Histological analysis—immunofluorescence (IF) staining
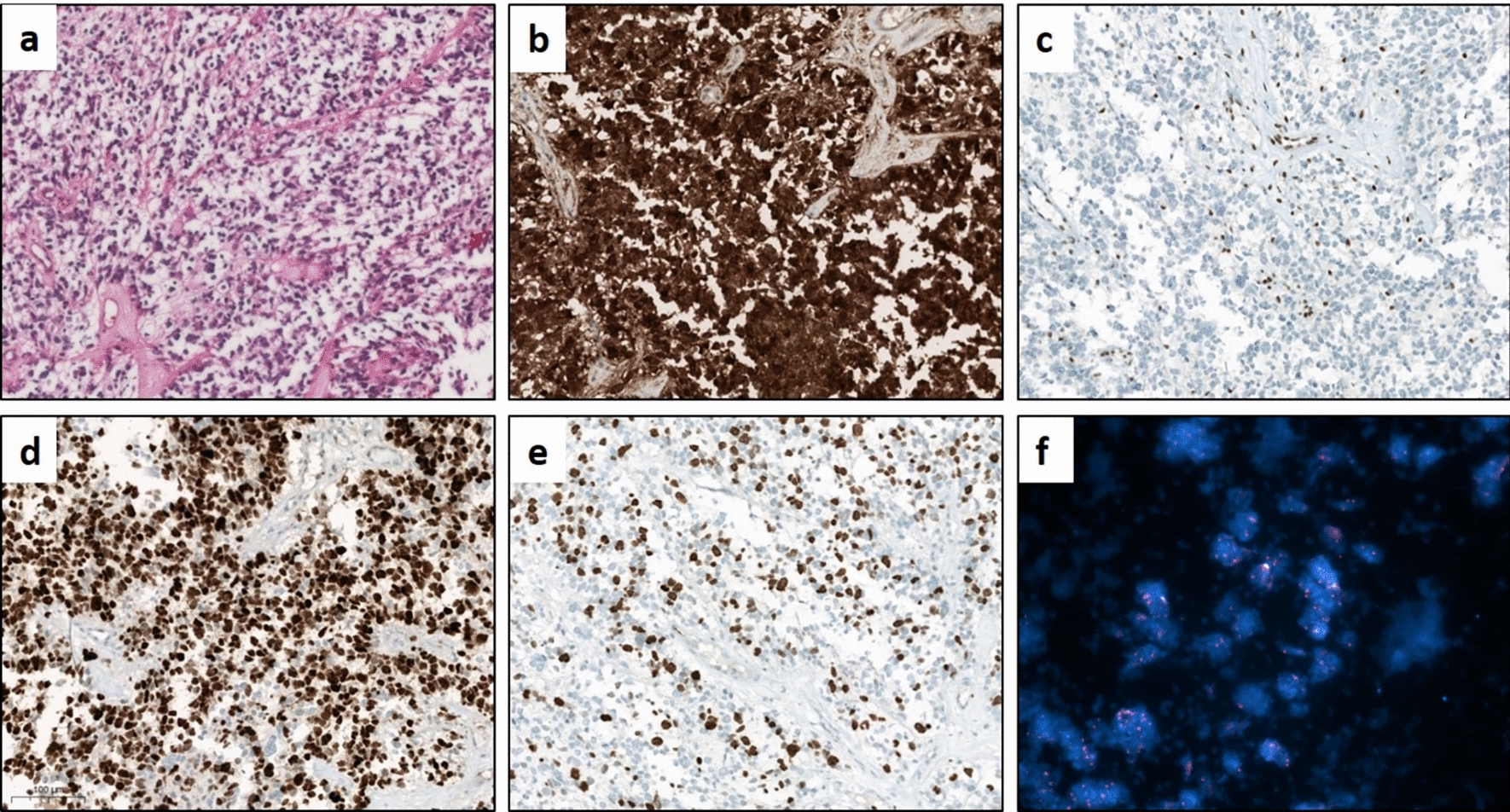
The slide was fixed with 95% methanol at − 20 °C for 10 min and washed twice with PBS for 5 min. The tissue was then surrounded by a hydrophobic barrier pen. The slide was treated with protease at 40 °C for 8 min and then washed 3 times with PBS for 1 min. To block the non-specific binding, the slide was incubated with a blocking buffer (5% FBS and 0.1% TWEEN 20 (Merck, P1379, USA) in PBS) for 60 min at RT. After blocking, the slide was incubated with the 200 μL primary antibodies diluted in blocking buffer at 4 °C overnight (12–16 h). Following the primary antibody staining, the slide was washed twice with washing buffer (0.1% TWEEN 20 in PBS) for 5 min. A secondary antibody diluted in a blocking buffer was then added to label the primary antibodies. The slide was incubated with 200 μL secondary antibodies at RT for 1 h and was washed twice with washing buffer for 5 min. The slide was then incubated with 200 μL Hoechst solution (1.25 μg/mL Hoechst 33342 in PBS) (Hoechst 33342: Thermo Fisher, 1871965, USA) to label cell nuclei at RT for 5 min. Following Hoechst staining, the slide was washed twice with PBS and let dry. Finally, a 10 μL mounting medium was dropped on each tissue slice, and the slide was covered with a coverslip. The image was viewed and captured by confocal microscopy (ZEISS, LSM780). Antibodies used and the dilution ratio are as follows: CD68 (Bio-Rad, MCA1957GA, USA; 1:200), GFAP (Abcam, ab7260, USA; 1:400), APOE (Sigma, 178479, USA; 1:1000).
Histological analysis—in situ hybridization staining
The slide was removed from − 80 °C and let air dry at RT for 2 h. The slide was fixed with 4% PFA at 4 °C for 20 min and washed twice with PBS for 1 min. The tissue was then surrounded by a hydrophobic barrier pen. In situ hybridization staining was performed with ViewRNA ISH Fluorescence Tissue Assay kit (Thermo Fisher, QVT0600C) according to the manufacturer’s instructions. The slide was treated with 100 μL protease at 40 °C for 10 min and then washed 3 times with PBS for 1 min. The slide was re-fixed with 4% PFA at RT for 5 min and then washed 3 times with PBS for 1 min. The slide was then incubated with a 100 μL Apoe target probe (Thermo Fisher, VX-06, assay ID: VB1-10244-VT) at 40 °C for 2 h and then washed 3 times with wash buffer for 2 min. Following target probe hybridization, the slide was incubated with 100 μL pre-amplifier solution at 40 °C for 30 min and then washed 3 times with wash buffer for 2 min. After pre-amplifier hybridization, the slide was incubated with 100 μL amplifier solution at 40 °C for 30 min and then washed 3 times with wash buffer for 2 min. After amplifier hybridization, the slide was incubated with 100 μL label probe solution at 40 °C for 30 min and then washed 2 times with wash buffer for 2 min. Finally, 10 μL Prolong antifade mountant with DAPI (Invitrogen, P36931, USA) was dropped on a tissue slice and the slide was covered with a coverslip. For ISH combined with IF staining, after label probe hybridization and washing, the slide was blocked with 100 μL blocking buffer (4% FBS, 1% goat serum, and 0.1% TWEEN 20 in PBS) at RT for 1 h. The slide was then incubated with primary antibody diluted in the blocking buffer overnight at 4 °C (12–16 h). Following the primary antibody staining, the slide was washed twice with washing buffer (0.1% TWEEN 20 in PBS) for 5 min. A secondary antibody diluted in a blocking buffer was then added to label the primary antibodies. The slide was incubated with 100 μL of secondary antibodies at RT for 1 h and was washed twice with PBS for 5 min. Finally, 10 μL Prolong antifade mountant with DAPI was dropped on a tissue slice, and the slide was covered with a coverslip. The image was viewed and captured by confocal microscopy.
Brain tissue dissociation for flow cytometry analysis
Mice were anesthetized with a Zoletil–Rompun solution. Intracardiac perfusion with PBS (15 mL) was performed to eliminate blood contamination. The brain was removed from the skull following the removal of the cerebellum and olfactory bulb and then dissected into three parts: the left hemisphere, the right hemisphere, and the tumor. The tumor tissue was removed from the brain and placed in a 6 cm dish prefilled with 4 mL cold PBS, while the brain tissue was transferred into a 50 mL tube prefilled with 2 mL cold DMEM and kept on ice until the next step. For the brain tissue dissociation, the brain tissue with 2 mL DMEM was transferred into the pre-cooled Tenbroeck Grinder (DWK life science, 885000-0015, Mainz, Germany). The tissue was homogenized by gently rotating the pestle while moving the pestle up and down 20 times. After Tenbroeck homogenization, an additional 3 mL DMEM was added into the grinder to a final volume of 5 mL. The sample was then transferred into a 50 mL tube and then homogenized with a syringe (5 mL) and needle (21-gauge) 10 times. The homogenate was then filtered through a 70 μm cell strainer and centrifuged for 5 min at 400 rcf at room RT. After centrifugation, the pellet was resuspended with 1 mL 1X RBC lysis buffer (Thermo Scientific, 00-4300-54, CA, USA) and kept on ice for 5 min. 2 mL PBS was then added to stop the reaction, and the sample was centrifuged for 5 min at 400 rcf at RT. The whole homogenization process was kept on ice to reduce cell death. Percoll solution (GE Healthcare, 17-0891-01, Uppsala, Sweden) was used to remove myelin. A stock solution of isotonic Percoll (SIP) was prepared by mixing 9 mL Percoll solution and 1 mL 10X HBSS (Gibco, 14,185). The cell pellet was resuspended in 4 mL 37% SIP (3.7 mL SIP + 5.3 mL 1 × HBSS) and centrifuged for 20 min at 1200 rcf at RT without brake. After centrifugation, the myelin at the top of the gradient was removed by pipette aspiration. The cell pellet at the bottom of the centrifuge tube was collected.
Tumor tissue dissociation for flow cytometry analysis
For the tumor tissue dissociation, the tissue was dissected into small pieces with a surgical blade. The tissue was transferred into a 15 mL tube and centrifuged for 5 min at 400 rcf at RT. After centrifugation, tissue was digested by 4 mL diluted ACCUMAX™ solution (STEMCELL TECHNOLOGY, 07921, USA; diluted ACCUMAX™ solution: 1 mL ACCUMAX™ + 4 mL 1X PBS) with gentle shaking for 60 min at RT. The homogenate was then filtered through a 70 μm cell strainer and centrifuged for 5 min at 400 rcf at RT. After centrifugation, the pellet was resuspended with 1 mL 1X RBC lysis buffer and kept on ice for 5 min. 2 mL PBS was then added to stop the reaction, and the sample was centrifuged for 5 min 400 rcf at RT.
Flow cytometry analysis
To block the Fc receptor, cell pellets were resuspended with blocking buffer and kept on ice for 15 min (blocking buffer: 0.2% FcR blocking reagent (BD, 553,142, USA) and 1% goat serum (Thermo Scientific, 16,210,064) in 1X PBS). To label the surface antigens (CD45, CD11b), 100 μL of cell suspension was added into 100 μL staining buffer containing antibodies (staining buffer: 1% goat serum in 1X PBS) and kept on ice for 30 min. After incubation, 1 mL staining buffer was added to wash out the antibodies, and the cells were centrifuged for 5 min at 400 rcf at RT. Antibodies used to label surface antigens are as follows: CD45 (BD, 552,848; 1:200), CD11b (BD, 550,993; 1:200). Subsequently, cells were fixed and permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD, 554,714) to label the intracellular antigen (APOE). Cell pellets were first vortexed and resuspended with 200 μL fixation/permeabilization solution for 20 min on ice. After fixation, 1 mL Perm/Wash Buffer was added to wash out the fixation buffer, and cells were centrifuged for 5 min at 1000 rcf at RT. Subsequently, cell pellets were resuspended with 200 μL Perm/Wash Buffer containing antibodies and incubated on ice for 30 min. After incubation, 1 mL Perm/Wash Buffer was added to wash out the antibodies, and the cells were centrifuged for 5 min at 1000 rcf at RT. After centrifugation, cell pellets were resuspended with PBS and stored at 4 °C in the dark until flow cytometry analysis. The antibody used is as follows: APOE (NOVUS, NB110-60531PE, USA; 1:200). Flow cytometry analysis was performed using BD FACSAria III flow cytometer.
Statistics
The image was analyzed by QuPath software [17]. The flow cytometry data were analyzed by FlowJo V10.6.2. Statistical significance was determined using a two-tailed unpaired Student’s t-test, with significance reported as p values < 0.05. * represents p ≤ 0.05, ** represents p ≤ 0.01, and *** represents p ≤ 0.001. All calculations and quantitative plots were performed using GraphPad Prism 8.0 (GraphPad Software Inc.).










Comments (0)