
Remember me









The coexistence of three histopathologically distinct lesions in the sellar region is extremely rare. Most publications identify at least two pathologies, such as a pituitary adenoma with another lesion [1,2,3,4,5,6] or hypophysitis associated with the rupture of RCC [7,8,9,10,11,12,13,14,15]. Following PRISMA guidelines for systematic review, until Jan 25, 2024, we searched the medical literature reporting cases with more than two sellar lesions written in English or the Spanish language using the PubMed electronic database and the keywords "Sellar lesions OR Sellar tumor lesions" AND "Rathke's cleft Cyst OR craniopharyngioma neoplasm" AND "pituitary adenoma OR pituitary neoplasm" OR "Xanthogranulomatous Hypophysitis OR Hypophysitis." We also performed manual searches of additional relevant studies in Scopus, identifying only two reported cases of three histopathologically distinct lesions (Table 1, Online Resource 1) in the Sellar region; both cases were also reported in female patients aged 71 and 35, respectively. The first of them reported the presence of a non-functioning adenoma accompanied by the presence of lymphocytic hypophysitis and primary pituitary lymphoma. In contrast, the second reported a growth hormone-producing microadenoma accompanied by the presence of lymphocytic hypophysitis and RCC [16, 17]. However, here, we describe a case that presents this for the first time: PA, RCC, and the rare XGH concurrently.
Table 1 Triple sellar collision lesions that are reported in the literatureThe pituitary gland is the primary endocrine organ regulating the body's hormonal balance. Its embryonic development involves the intricate interaction of different tissues. The anterior pituitary originates from the oral ectoderm (Rathke's pouch), while the posterior pituitary develops from the neural ectoderm of the diencephalon during weeks 3–4 of fetal development [18]. It is located in the hypophyseal fossa (sella turcica), a mid-skull region with great structural complexity due to the presence of surrounding structures, such as the internal carotid artery, cavernous sinus, optic chiasm, hypothalamus, and the pituitary gland [19, 20]. Sellar lesions are any neoplastic or tumor-like mass originating in any of the components that make up this region [20]. They represent frequent lesions, an incidental finding in up to 10–30% of MRI scans indicated for another cause [21, 22], and a finding in 10% of autopsies [23].
In most cases, they usually present as single lesions; however, in exceptional conditions (< 2% of cases), they present concomitantly as a collision tumor [5]. Clinical manifestations are nonspecific and are usually secondary to mass effects on adjacent anatomical structures or hormonal abnormalities [24]. The primary differential diagnoses for suprasellar lesions include pituitary adenomas, Rathke’s cleft cysts, craniopharyngiomas, lymphocytic hypophysitis, metastatic tumors, and less common entities, such as granular cell tumors or abscesses. Given the limited specificity of imaging studies, an accurate diagnosis and appropriate management require a comprehensive evaluation that integrates clinical, biochemical, and histopathological findings [25, 26].
Pituitary adenomasPAs are the most common sellar lesion, accounting for up to 90% of cases in adults and representing 10–15% of all brain tumors [25, 26]; they are benign clonal neoplasms of the neuroendocrine cells of the adenohypophysis [27]. They are classically classified as micro (< 10 mm diameter) and macroadenomas (> 10 mm diameter), as well as hormone-active (functional) and hormone-inactive (non-functional) adenomas depending on whether they are associated with hormone production [20, 28], 70% of them being hormonally active [29]. On MRI, the presence of a homogeneous isointense image on T2 and T1 is typical [30], which contrasts with what was observed in our case, where a heterogeneous image was presented, possibly explained by the size of the lesion. However, when performing a contrast-enhanced MRI, enhancement of the lesion was observed, constituting a gold standard for diagnosing PA [29].
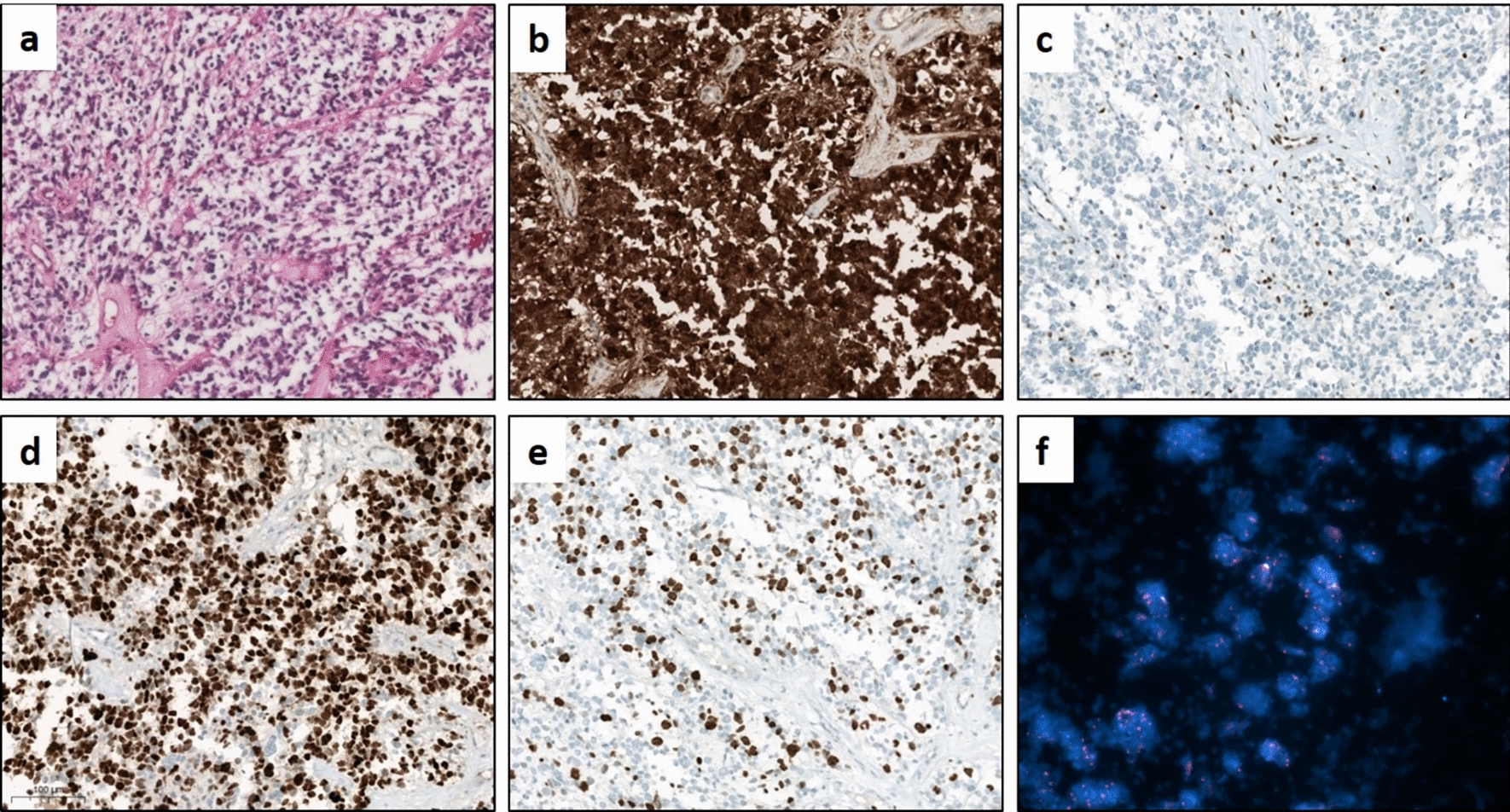
Histologically, these lesions are characterized by rounded epithelioid cells with granular cytoplasm, round nuclei with finely dispersed chromatin, and multiple distinct nucleoli [27]. The above corresponds to what was found in our patient's mass, where we observed foci of the lesion characterized by acute and chronic inflammatory responses, pseudostratified ciliated columnar epithelium, foamy macrophages, cholesterol clefts, and dystrophic calcifications, along with non-functional pituitary adenoma with no hormone activity. (Fig. 2D). In our case, despite finding increased free thyroxine in the endocrinological profile, we did not find immunoreactivity to any of the pituitary hormones, and given that the thyroid-stimulating hormone values were found within normal ranges, the most probable cause for this endocrinological finding would be an episode of asymptomatic thyroiditis [31]. According to the World Health Organization (WHO), adenoma classification requires identifying transcription factors [28]. Therefore, and as a limitation of our case, we cannot fully establish a null cell adenoma since 1/5 of clinically non-functioning adenomas are negative for all anterior pituitary hormones but positive for the steroidogenic factor-1 (SF-1), being indicative of a gonadotroph lineage [32, 33].
Rathke cleft cystRCCs are non-neoplastic cystic lesions arising from remnants of Rathke's pouch, formed between the anterior and posterior lobes of the pituitary gland. Symptomatic cases are rare, and their epidemiology is uncertain [34]. Macroscopically, the cyst is encapsulated in a delicate membrane, and the contents can be diverse, with 65% having a proteinaceous content formed by hemosiderin, cholesterol, mucin, and other proteins [35]. The content of the cyst is of interest, as variations have been observed in the clinical and imaging findings, cysts with proteinaceous content being the ones that are associated with the presence of symptoms such as headache [36, 37], which coincides with our case, where the presence of a proteinaceous content associated with symptomatology was present. The MRI signal depends on the cyst content; on T1-weighted images, RCCs may present both hypo- or hyperintense; on T2-weighted sequences, about 70% appear hyperintense, while 30% are iso-to-hypointense. These are characteristics of an intracystic non-enhancing nodule and enhanced rim of compressed pituitary surrounding the cyst after contrast administration [20, 34]. In the present case, the imaging findings do not comply with the above; however, it should be noted that in the presence of a collision tumor concomitant to a PA, it is difficult to establish the differential diagnosis by imaging [2, 5]. In fact, it was observed that of a total of 39 cases, only 15 presented two different magnetic MRI signals that allowed to suspect both lesions [6].
Histologically, the cyst's wall is formed by a simple or pseudostratified cuboidal or columnar epithelium, which expresses EMA, a transmembrane glycoprotein typically expressed in luminal epithelial cells that provides barrier functions. The epithelium of the cyst also contains ciliated cells and goblet cells, the last responsible for the production of the mucinous material that accumulates inside the cyst [34]. In the present case, we found remains of the cyst's wall, which is most likely due to a spontaneous cyst rupture. It has been observed that the cyst wall undergoes cyclical changes of inflammation that cause bleeding and subsequent rupture and degeneration of the cyst and can even trigger xanthogranulomatous changes [12, 13, 38]. The above indicates that RCC, xanthogranulomas, and craniopharyngiomas constitute a spectrum of the same entity [39]. Nonetheless, rupture of the cyst could be favored by other mechanisms, including increased intra-cystic pressure due to dysregulation of content secretion–reabsorption, decreased cyst wall thickness related to chronic inflammatory changes, or mass effect exerted by pressure with other structures [9, 40, 41].
HypophysitisHypophysitis is the acute or chronic inflammation of the pituitary gland and is classified as primary (idiopathic) or secondary to sella turcica and parasellar region lesions, systemic diseases, or drugs [25]. It is a rare disease compared to other pituitary lesions, with an estimated prevalence of 0.2–0.88% [42]. Histologically, it can be classified as lymphocytic (the most common form), granulomatous, xanthomatous, IgG-4 related, necrotized, and mixed forms such as XGH (the least frequent) [42, 43]. In MRI studies, stalk thickening and homogenous enlargement of the pituitary gland are typically observed; however, imaging is not specific [43].
The granulomatous form is typically idiopathic or secondary to systemic diseases, such as tuberculosis or Langerhans disease [43]. Histologically, it is characterized by numerous multinucleated giant cells, calcium deposits, lymphocytes, and histiocytes with granuloma formation [11]. The above is characteristic of a response to a foreign body [44]. On the other hand, the xanthomatous variety is frequently secondary to local processes, commonly the rupture of an RCC [13]. Histologically, it presents CD68-positive foamy cells, cholesterol clefts, and hemosiderin deposits [45]. Here, we report a case of an extremely rare subtype of hypophysitis called XGH, exhibiting xanthomatous and granulomatous features characterized by the presence of cholesterol clefts, lymphocytes, plasma cells, hemosiderin deposits, fibrosis, foreign body giant cells, eosinophilic remnants, caseating and necrotizing granuloma, accumulation of foamy macrophages, and granuloma formation [46, 47]. This variety of hypophysitis may be primary with an autoimmune etiology or secondary as a reactive degenerative response to an epithelial lesion or as a part of a multiorgan systemic [46]. This case is particularly notable, because it presents alongside a PA and an RCC, suggesting a complex network of pathophysiological mechanisms. The concurrent development of these three lesions may indicate a shared inflammatory or immune response within the hypophyseal fossa, possibly driven by the local microenvironment or a systemic predisposition to multiple pathologies. Understanding the histopathological interactions and triggers leading to such unusual presentations could provide deeper insights into pituitary disease mechanisms and guide more effective diagnostic and therapeutic approaches.
Explaining the coexistence of a collision tumorMultiple theories have been proposed to explain the existence of collision tumors in the sellar region [1, 5], particularly the coexistence of PA with RCC is the most frequent finding. Furthermore, prolactinomas or somatotroph lineage adenomas are more common than non-functioning adenomas, such as the one presented in this case [1]. However, none of those theories explain the presence of all collision tumors, so their existence as a coincidence remains feasible. Therefore, our objective is to discuss the interrelationship between these lesions rather than explaining their same origin.
The development of the pituitary gland is a complicated process that involves the function of various genes and signaling pathways in the first 4–8 weeks of gestation (Fig. 3A, B) [27], during which, occur the formation of the two main lobes of the gland and closure of Rathke's cleft in the pars intermedia. The cause of the incomplete closure of this cleft, which would give rise to the formation of RCC due to the accumulation of material secreted by its wall, has not yet been identified (Fig. 3C, E)[2, 40]. Specifically, no mutations in the genes or signaling pathways involved in the development of the pituitary gland have been related to this phenomenon or the development of PAs. Interestingly, in addition to PAs with a familial predisposition, it has been observed that mutations in the GNAS and USP-8 genes are the only significant molecular mechanisms in sporadic somatotroph and corticotroph PAs, probably by favoring the mitogenic signal or by deregulation of the cell cycle, respectively [27, 48].
Fig. 3
Proposed pathological mechanisms of the possible relationship between pituitary adenoma, Rathke cleft cyst, and xanthogranulomatous hypophysitis. Development of the pituitary gland at (A) 4 and (B) 8 gestation weeks, as well as in (C) the adult stage. D, F, I Possible origins of the pituitary adenoma. E Development of the Rathke cleft cyst after the accumulation of the secretion of cellular remnants in the Rathke's cleft. G, H Possible origins of xanthogranulomatous hypophysitis. This figure was created with BioRender.com
Complementary studies in animal models have identified that mutations in the Prop-1 gene are associated with a higher incidence of PAs or RCC independently and without increasing the frequency of a collision tumor [49]. However, we can suggest that there is an interaction of RCC content that could stimulate the lactotrophic cells of the adenohypophysis and cause the development of an adenoma (Fig. 3F) [50].
On the other hand, cysts with mucinous content tend to superinfection [35] and consequent inflammation and rupture, which would cause a constant leak of cyst material (Fig. 3G). The continuous exposure of the pituitary epithelium to the cyst's contents would cause an inflammatory response [7, 9], in the present case of the foreign body type, mediated by mucin and hemosiderin [14, 39]. Furthermore, the presence of a PA could stimulate or trigger this inflammatory response, since it has been observed that PAs secrete pro-inflammatory cytokines (IL-1, IL-2, and IL-6) associated with the development of hypophysitis (Fig. 3H)[51] or even the inflammation could be secondary to silent apoplexy of the PA [39]. Finally, this chronically maintained inflammatory response could cause epithelial hyperplasia and squamous metaplasia of the pituitary gland and subsequent development or growth of a PA (Fig. 3I) [16]. In this case, the coexistence of a PA, RCC, and XGH highlights a complex interplay of pathological mechanisms. The RCC may have initiated inflammation that led to PA development, and pro-inflammatory cytokines from the PA could have exacerbated this inflammation, facilitating XGH formation.
In addition, the continuous exposure of pituitary tissue to the leaking RCC content could have perpetuated the inflammatory response. This sequence of events implies that each lesion influences the development and evolution of the others. Understanding these dynamics is crucial for improving the diagnosis and treatment of complex pituitary lesions.
Comments (0)