
Remember me


In this study, the next-generation sequencing technique was used to examine the molecular basis of hearing loss in a group of 48 Polish patients with clinically diagnosed, not DFNB1, hearing loss. To our best knowledge, it was so far the largest Polish group of patients with NSHL analyzed with the NGS technique. Molecular diagnosis was established for 39.6% of the patients. Among thirty-three identified pathogenic/potentially pathogenic variants, six are novel: p.Gly1326Val (STRC), p.Pro104ThrfsTer2 (MYO6), p.Tyr186Ter (GATA3), p.Ile1584SerfsTer12 (MYO15A), p.Pro559Leu, and p.Glu542del (CDH23).
The similar results of molecular diagnosis established for 39% (440/1119) of HL American patients were shown using the OtoSCOPE platform (version 4 and 5), with no pre-screening (Sloan-Heggen 2015). In the NGS study (OtoSCOPE v5), performed in 302 HL patients of Iranian origin with excluded biallelic pathogenic variants in the GJB2 gene, the frequency of established molecular diagnosis was higher (67%) (Sloan-Heggen et al. 2016). However, these patients showed a high rate of consanguineous marriages. In a Dutch study of 200 HL patients using the NGS technique (120 selected HL genes) the detection rate was 33.5%, but DFNB1 patients were also included (Zazo Seco 2017). Shearer and Smith (2015) performed a retrospective analysis of the results of using the NGS technique to detect variants responsible for hearing loss from 20 publications (603 HL patients in total). The average detection rate was 41%. Differences in detectability from 10 to 83% resulted from the selection of the study group (patients with NSHL or SHL), the type of panel used (different number of tested genes), the performance of additional preliminary analysis, and CNV detection. However, the exact comparison of the results between the studied HL populations is difficult due to the large number of genes associated with HL, the different size of the study groups, and the lack of a unified analysis algorithm (Shearer and Smith 2015).
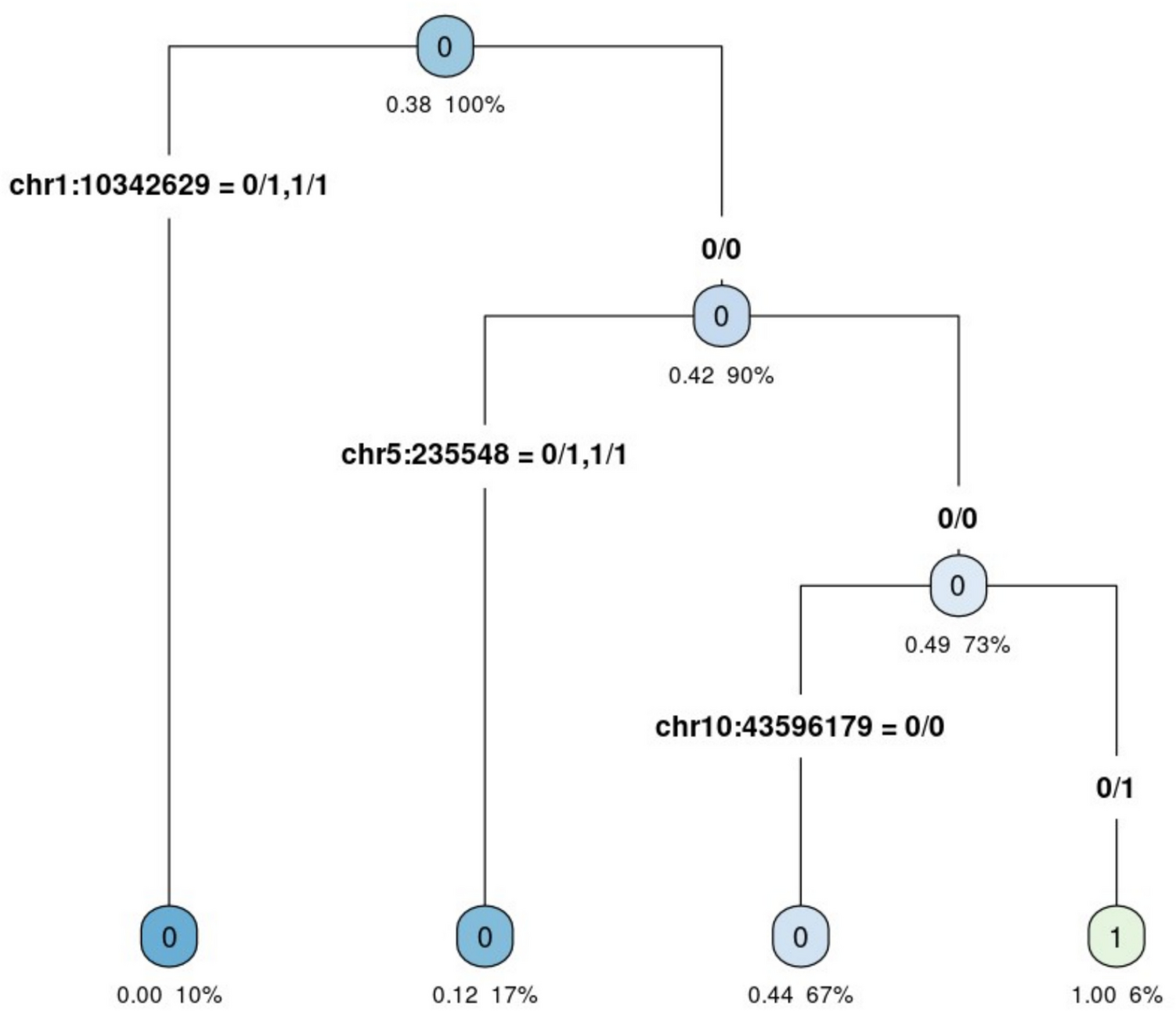
The presented results indicate the complexity of the molecular basis of hearing loss in Polish patients with NSHL. Causal variants were identified in thirteen different genes. Only in STRC, USH2A, and MYO7A genes, pathogenic variants were found more than once, with STRC gene deletion being the most common molecular defect. The molecular heterogeneity of HL was also present in other populations. In the study of the 207 NSHL French families, the molecular basis of HL was determined in 48% of the examined patients. Half of them have DFNB1A hearing loss. For the remaining cases, causal variants were identified in 19 genes, with STRC gene in the first place (Baux 2017). Studies of NSHL in Spanish patients revealed pathogenic variants in 16 genes (GJB2/GJB6, OTOF and MT-RNR1 genes were prescreened) (Cabanillas 2018). In 45 out of 58 Chinese HL patients with NSHL and SHL, pathogenic variants were identified in as many as 24 genes; however, with a predominance of variants in the SLC26A4 (35.80%) and GJB2 (12.35%) genes (Sun 2019). The complexity of the molecular basis of HL is evident even within single families in this study, where pathogenic variants in two different genes are responsible for hearing loss within one family: the TSO_8 family, these are USH2A and GJB2 genes (Niepokój 2018), the family P12, the GJB2 and MYO15A genes (Fig. 1), and in the family P_28, the GJB2 and STRC genes (Fig. 2).
Fig. 1
Family P_12—causative pathogenic variants in two different genes in one family. Symbols: empty figures—unaffected individuals, black figures—hearing loss, half-black—unaffected carrier, circle—female, square—male; GJB2: NM_004004.6:c.35del (p.Gly12ValfsTer2); c.313_326del (p.Lys105GlyfsTer5); MYO15A: NM_016239.3:c.4749del (p.Ile1584SerfsTer12); c.8183G > A (p.Arg2728His)
Fig. 2
Family P_28—causative pathogenic variants in two different genes in one family. Symbols: black figures—hearing loss, half-black—unaffected carrier, circle—female, square—male, GJB2: NM_004004.6:c.35del (p.Gly12ValfsTer2); c.269 T > C (p.Leu90Pro)
In most cases (74%), pathogenic variants detected in our study group were responsible for the autosomal recessive hearing loss (genes: STRC, USH2A, MYO7A, OTOA, OTOG, MYO15A, CDH23 and HARS2), and 26% for AD hearing loss (MYO6, GATA3, MITF, ACTG1, PTPRQ genes). In total, thirty-three causative variants were identified, with the majority (60%) identified only once among the patients of the study group (66% if we also consider homozygous pathogenic variants). Six of the thirty-three identified pathogenic variants are not described in the HGMD Professional and ClinVar databases. The obtained high percentage of loss-of-function (LoF) pathogenic variants among the total causal variants in the study group (63,6%) is alike to the data published by Azaiez et al. (2018) in a retrospective analysis of the variants present in the Deafness Variation Database: 50% of all registered pathogenic variants. In the cases of known pathogenic variants described earlier in literature, their role in the pathogenesis of hearing loss was beyond doubt. It is worth noting that the p.Thr277Ile pathogenic variant in the ACTG1 gene in HL patient P_1 was unknown at the time of its identification. It was described by Liu and coworkers (2019) a three-generation Taiwanese family with post-lingual, progressive hearing loss, where both the symptoms of the disease and the pattern of inheritance were consistent with our study data.
In our study, the largest percentage of the thirty-three identified causal variants were deletions (39%), and among them, the STRC gene deletions are the most common (69% of all identified deletions in the study group). Thus, our research confirms the high frequency of STRC gene deletion in Polish patients with clinically diagnosed NSHL.
STRC gene deletions play a significant role in the pathogenesis of hearing loss (Yokota 2019). Depending on the size and selection of the study group, deletions of the entire STRC gene are a cause of hearing loss of 2% in Danish HL patients (Zazo Seco 2017), 11% in both American (Shearer 2014) and German (Back 2019) HL patients. According to Markova et al. (2018) in the Czech population, deletions of the entire STRC gene are the second, after pathogenic variants in the GJB2 gene, cause of NSHL (13,5%). Shearer et al. (2014) analyzed the CNV variants in HL patients and out of 267 subjects for whom a molecular cause of hearing loss was established, fifty had CNV changes in 16 genes (the STRC gene deletions predominated). Knijenburg et al. (2009) estimated the overall incidence of hearing loss caused by pathogenic variants in the STRC gene (DFNB16) at 1:16,000, and according to Hoppman et al. (2013), the carrier frequency of STRC gene rearrangements exceeds 1% of the general population. The high frequency of rearrangements in this gene is due to a tandem duplication at the locus 15q15.3 where the gene is located (Avidan 2003).
In the study group, we have identified pathogenic/potentially pathogenic variants in the genes responsible for SHL, which in consequence changed the clinical diagnosis (36,8% of genotyped HL patients). This may be a result of the relatively early age of the patients (average age was 21 years). In cases of SHL with gradually and not simultaneously developed symptoms, the correct clinical diagnosis may be significantly delayed. Patients with Usher syndrome are born with HL; however, a loss of vision progresses with age. The first vision-loss symptoms may arise at the age of 20–30 years. Also, in Perrault syndrome associated with gonadal dysgenesis or premature ovarian failure in women, the correct diagnosis may be delayed (men with this syndrome are fertile). The DIS syndrome associated with HL and male infertility is usually diagnosed in the procreative age when fertility problems become apparent (HL is present from birth). In syndromic HL, varying severity of syndromes has also been observed. In Waardenburg syndrome associated with pathogenic variants in the MITF gene, HL occurs in 77–80% of patients, heterochromia iridis in 42–54% of patients, and the characteristic white lock only in 16–23% of patients (Milunsky 2001). Similarly, in HDR syndrome, characterized by a triad of symptoms (HL, hypoparathyroidism, and renal developmental disorders), the clinical picture is diverse. Hearing loss is the most common, while hypoparathyroidism and kidney disorders occur in varying degrees of severity (Bernardini 2009).
Identification of an extensive deletion in both the STRC gene and the CATSPER2 gene in TSO_7 and TSO_16 patients resulted in a change of diagnosis towards DIS syndrome.Footnote 1 Identification of pathogenic variants in the genes USH2A (patients TSO_8 and TSO_14), MYO7A (patients P_18 and P_26), and in the CDH23 gene (patient P_23) suggests the diagnosis of Usher syndrome.
However, it should be noted that when pathogenic variants are identified in the MYO7A and CDH23 genes, the situation is more complicated, as these genes are responsible for both NSHL and Usher syndrome. For example, pathogenic variants in the MYO7A gene cause Usher syndrome (recessive inheritance) and isolated hearing loss (both recessive and dominant inheritance). Therefore, the identification of the causative genotype does not provide answers to clinical questions. In particular, MYO7A missense variants were reported both in syndromic and nonsyndromic disorders (Watanabe et al. 2024).
The problem of genetic pleiotropy and a pattern of inheritance also concerns other hearing loss genes, such as GJB2, PAX3, EDNRB, and COL11A2, that are causative for various clinical entities inherited in autosomal dominant and recessive modes. The solution may be to carry out variant segregation with the disease in the family. Therefore, variant segregation in the family and other additional tests and clinical evaluation may be crucial to draw final conclusions and set the diagnosis.
The change in clinical diagnosis has also affected patients TSO_15 (HDR syndrome), P_2 (Waardenburg syndrome), and P_8 (Perrault syndrome). Similar cases have also been described for NSHL patients from other populations. In studies of Baux et al. (2017) in 16% of patients with NSHL, pathogenic variants were identified in Usher syndrome genes. The Sloan-Heggen et al. (2015) showed that applying the NGS enabled the reevaluation of the clinical diagnosis from NSHL to SHL (Usher syndrome, Pendred syndrome, Charcot-Marie-Tooth disease, Chudley-McCullough syndrome, and Jervell-Lange-Nielsen syndrome) in 8% of diagnosed patients. In the case of 21 NSHL Spanish patients, the diagnosis was changed to Waardenburg syndrome, Usher syndrome, HDR syndrome, and CHARGE syndrome in patients (28,6%) with an NGS established molecular diagnosis (Cabanillas 2018).
The lack of molecular diagnosis in twenty patients (almost 42%) may be due to several reasons including reasons other than genetic causes of HL. A molecular defect in unexamined regions of the genes (introns, regulatory regions) as well as the presence of pathogenic variants in genes not included in the used panels.
Our studies indicated a heterogeneous genetic basis of the hearing loss of Polish patients. They revealed a significant contribution of the STRC gene deletion in the pathogenesis of NSHL. Additionally, the size of the deletion at the 15q15.3 locus draws attention to the problem of male infertility coexisting with hearing loss (DIS syndrome). The application of the NGS technique enables detecting cases of syndromic HL that have not been detected during a medical examination. Lack of clinical diagnosis often results from the specificity of these syndromes, the age of onset of symptoms, or the degree of their expression. Therefore, the use of the NGS technique should be an integral part of the HL diagnosis (Skarżyński 2021). If the molecular result suggests a change in the clinical diagnosis, it allows providing the appropriate medical care early enough.
The above cases illustrate both the complexity of the molecular basis of hearing loss and draw attention to the difficulties that may arise in the diagnostic process. In this context, it is clear how important it is to use an appropriate diagnostic algorithm and perform a variants segregation analysis in a family (Zhou 2012).
Supplementary information.
Comments (0)