
Remember me
The proband was an 11-year-old girl who was born at 38 weeks gestational age to healthy nonconsanguineous parents and weighed 2724 g. At the age of one year, she was referred to the Division of Ophthalmology, National Center for Child Health and Development (NCCHD) for a thorough examination of left eye strabismus and bilateral ptosis. She also had a history of infant gastrointestinal allergy. Her parents and elder brother had no notable diseases.
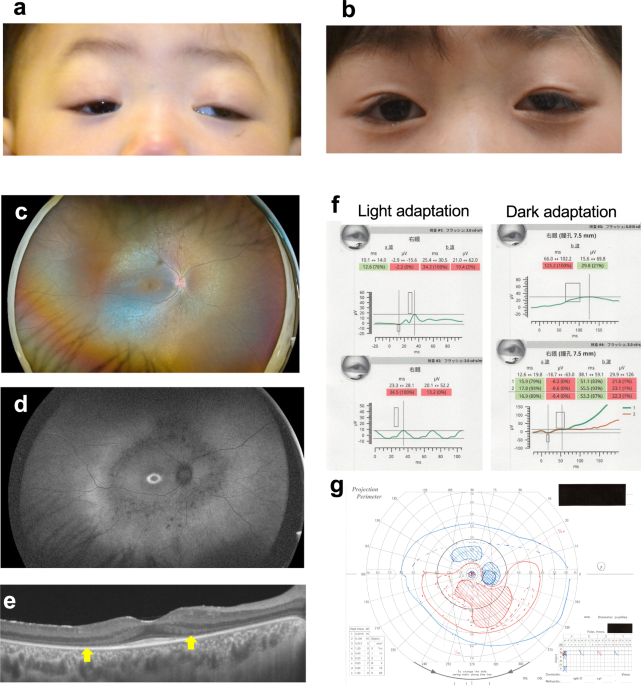
The initial ocular examination of the patient at the age of one year revealed congenital bilateral ptosis and left exotropia and hypertropia due to dissociated vertical deviation (Fig. 1a), but she maintained binocular fixation by elevating her chin. There were no abnormal findings in either the anterior segment or the fundus of either eye. No specific neurological or systemic abnormalities were identified. She underwent strabismus surgery for left exotropia at 2 years and 9 months of age and frontalis suspension for bilateral ptosis at 4 years and 9 months of age. At the age of 5 years, she achieved orthotropia (Fig. 1b) with coarse stereoacuity of 3000 s of arc on the Stereo Fly Test. Thereafter, periodic examinations showed no significant changes. However, at 8 years of age, ophthalmoscopy showed bilateral degeneration of the retinas necessitating comprehensive and detailed ophthalmic examinations. Around age 9 years, the patient’s night blindness and photophobia became significant. Ultra-wide fundus photography and autofluorescence imaging (California, Optos Plc, Dunfermline, UK) revealed extensive retinal degeneration and hypofluorescence in the mid-periphery at age 11 years (Fig. 1c, d). Central retinal architecture was evaluated using swept-source optical coherence tomography (SS-OCT) (DRI OCT-1, Topcon, Tokyo, Japan), which showed bilateral significant thinning of the ONL and a diminution of the EZ except for the foveal area (Fig.1e). Using ffERG employing the RETeval system (LKC Technologies, Inc. Gaithersburg, MD, USA), we observed decreased cone responses and markedly reduced rod responses in both eyes (Fig.1f). These findings confirmed a definitive diagnosis of bilateral rod-cone retinal dystrophy. The best-corrected decimal visual acuity was 0.7 in the right eye and 0.5 in the left eye. Goldman perimetry showed constricted visual fields with large paracentric scotomas in both eyes (Fig.1g). Systemic re-evaluations revealed mild developmental delay and slightly shorter-than-normal stature, but there were no other CHARGE syndrome-like features; neither atresia of the choanae, heart defects, ear abnormalities, hearing loss, genital anomalies, nor the hockey stick sign were present. The patient’s mother had no ocular or systemic abnormalities, although she did prove to be a carrier of one of the CDK9 variants.
Fig. 1: The ophthalmic phenotypes of the patient.
a Photograph showing the bilateral ptosis, and left exotropia and hypertopia, seen in the patient at the age of 14 months. b Photograph showing orthotropia in the patient at the age of five years following surgical treatment to correct her bilateral ptosis and left exotropia. c Ultra-widefield fundus photograph showing extensive retinal degeneration with ossicle-like pigmentation in the mid-periphery in the patient at the age of 11 years. d Fundus autofluorescence photograph demonstrating a hyperfluorescent ring around the fovea, and hypofluorescence in the mid-periphery where retinal degeneration was prominent, in the patient at the age of 11 years. e Swept-source optical coherence tomography image of the posterior retina showing severe thinning of the outer nuclear layer and a diminished ellipsoid zone (arrows) except in the foveal area. The patient was 11 years old. f Full-field electroretinography using the RETevalTM portable device revealing reduced cone responses (LA 3.0 and 30 Hz flicker), and markedly decreased rod responses (DA 0.01) and mixed rod and cone responses (DA 3.0), in the patient at the age of 11 years. g Goldman perimetry showing constricted visual fields with large paracentric scotomas in the patient at the age of 11 years
Molecular studiesTo determine the genetic basis of the patient’s eye anomalies, genomic DNA was extracted from peripheral blood samples of the patient and her parents. Trio-based whole-exome sequencing was performed. Exome capture was carried out using the Twist Exome 2.0 (Twist Bioscience, South San Francisco, CA, USA), and sequencing was performed on a NovaSeq 6000 (Illumina, San Diego, CA, USA) with 150-bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed as described previously [7]. Using PLINK (v1.90; https://www.cog-genomics.org/plink/1.9/) [8], we calculated PI_HAT based on 4,225 variants from the trio samples together with 359 individuals from the 1000 Genomes Project. The PI_HAT values between the patient and each parent were 0.5 (Z1 = 1), thereby confirming both paternity and maternity. Read-depth-based copy number variation analyses were performed using the exome-hidden Markov model (XHMM) [9] and jNord [10]. After the filtering steps, we identified two rare missense variants (c.862G>A, p.A288T and c.961C>T, p.P321S) in the CDK9 (NM_001261.4) gene. The c.862G>A variant was maternally inherited, while c.961C>T was not found in either parent (Fig. 2a, b). Visualization with the Integrative Genomics Viewer (IGV) confirmed that c.862G>A and c.961C>T were present on different reads, indicating that they are in trans (Fig. 2c). The c.862G>A variant was previously reported by us as a causative variant in CHARGE syndrome-like retinal dystrophy [6]. The c.961C>T variant was considered to be a novel de novo variant that was absent from population allele frequency databases such as gnomAD v4.1.0 (https://gnomad.broadinstitute.org/; accessed on 16 May 2025) and 60KJPN (https://jmorp.megabank.tohoku.ac.jp/; accessed on 16 May 2025) [11], and was predicted to be pathogenic by multiple in silico prediction tools (Supplementary Table 1). The patients also possessed the c.633+1G>A variant of the PDE6C gene, which is known to cause inherited retinal dystrophy in an autosomal recessive manner [12]. However, since only a single allele of this variant was found in the patient’s DNA, and the patient’s phenotype was inconsistent with that typically associated with PDE6C variants, it was considered unlikely to be causative in our patient’s case.
Fig. 2: A novel CDK9 genetic variant in the patient.
a Pedigree of the patient’s family showing CDK9 variants. The CDK9 genotypes of the proband (II-2) and parents (I-1 and I-2) are shown. Arrow, proband; square, male; circle, female; black, affected individual; +, wild-type allele; *, DNA sample not available. b Electropherograms of CDK9 variants in the proband and parents. Partial CDK9 sequences from the proband (II-2) and parents (I-1, I-2) are shown. Variant positions are highlighted in yellow. The c.862G>A variant is maternally inherited, whereas the c.961C>T variant is absent in both parents, indicating a de novo origin in the patient. c IGV view of CDK9 variants in the proband showing that the c.862G>A and c.961C>T variants are located on different alleles (in trans). The c.862G>A variant is highlighted in light green and the c.961C>T variant in red. The allele carrying c.862G>A is outlined in green, and the allele carrying c.961C>T is outlined in red
As previously determined, the orthologous CDK9 genes in human and zebrafish are highly conserved in structure and amino acid sequence, and show an identical cyclin-binding motif (Fig. 3a). The amino acid residue A288 is located in the protein kinase catalytic domain of CDK9, but the P321S residue mutated in our patient lies outside this region in both species. To compare the kinase activities of the P321S and A288T variants, we prepared Flag-tagged recombinant versions of the wild-type (WT), P321S and A288T CDK9 proteins and expressed them in HEK293 cells. Interestingly, the kinase activities of both the P321S and A288T variants were reduced compared to the WT enzyme (Fig. 3b). To test the relative effects of these lower CDK9 kinase activities on gene expression patterns, we employed a NF-κB reporter gene assay. We observed that the levels of NF-κB activity induced by the three CDK9 proteins could be ranked as WT 100% > P321S 70% > A288T 50% (Fig. 3c). These results clearly show that the patient’s novel P321S variant impairs CDK9 kinase activity but not to the same extent as the A288T variant, which may result in a milder phenotype (Fig. 3d). Both variants were classified as variants of uncertain significance (VUS) according to the ACMG/AMP guidelines [13] refined by the ClinGen Sequence Variant Interpretation (SVI) Working Group [14,15,16] (Supplementary Table 1).
Fig. 3: Biochemical properties of the novel CDK9 variant.
a Diagram of the domain structure of the human and zebrafish CDK9 proteins. Numbers indicate amino acid positions from the N-terminus of human CDK9. Arrows indicate arginine (R) 225 substituted with cysteine (C), alanine (A) 288 substituted with threonine (T), arginine (R) 303 substituted with cysteine (C), and proline (P) 321 substituted with serine (S) (new variant in the patient). b Top: In vitro autophosphorylation assay of kinase activities of Flag-tagged human WT, P321S, and A288T CDK9 enzymes in the presence of cyclin T1. Data are the mean enzymatic activity rate ± SD of results obtained from one representative experiment in which each transfection was performed in triplicate. *p < 0.05, **p < 0.01 by Student’s t test. Bottom: Immunoblotting with anti-Flag antibodies to detect Flag-CDK9 proteins in the assay in the top panel. c NF-κB-dependent gene activity assay of HEK293 cells that were transfected with the NF-κB-Luc reporter plasmid and either Flag-tagged human CDK9 WT, P321S or A288T expression plasmids, plus CyclinT1, as indicated. After 24 h incubation, cells were harvested and luciferase activity was measured. Data are the mean values shown are the mean NF-κB activity ± SD of results obtained from one representative experiment in which each transfection was performed in triplicate. *p < 0.05, **p < 0.01 by Student’s t test. d Diagram of the proposed impact of pathological human CDK9 biallelic variants. Assuming that the kinase activity of wild-type CDK9 is 100%, the total kinase activities of the biallelic CDK9 variants A288T/P321S and A288T/R303C are about 60% and 30% of the WT value, respectively. A patient bearing A288T/P321S would show only retinal dystrophy, whereas a patient with A288T/R303C would exhibit both retinal dystrophy and CHARGE-like malformation syndrome
Comments (0)