Experimental methodsGlioblastoma cell culture
LN229 cells, a female human GBM cell line obtained from the American Type Culture Collection, were cultured as a monolayer in coated T-75 flasks (Nunclon Delta coated, Fisher) using DMEM/F12 Glutamax (Thermo Fisher) supplemented with 5% fetal bovine serum (FBS, Fisher) and 1% Pen/Strep (Thermo Fisher). Cells were maintained in a 5% CO2 incubator at 37 °C. Passage number in all experiments ranged from six to twelve.
Glioma 261 cells (GL261, obtained from the Division of Cancer Treatment and Diagnosis Tumor Repository of the National Cancer Institute), a male murine model of GBM, were cultured in monolayer in coated T-75 flasks (Nunclon Delta coated, Fisher) using RPMI-1640 (Sigma-Aldrich) supplemented with 10% FBS (Fisher), 1% L-Glutamine (Thermo Fisher), and 1% Pen/Strep (Thermo Fisher). Cells were maintained in a 5% CO2 incubator at 37 °C. Passage number in all experiments ranged from six to twelve.
Nf1−/−DNp53 male astrocytes (Generous Gift of Dr. Joshua Rubin, Washington University in St. Louis), a syngeneic murine model of GBM, were cultured in monolayer in coated T-75 flasks (Nunc Treated EasYFlasks, Fisher) using DMEM/F12 media (Gibco), supplemented with 10% FBS (Fisher) and 1% Pen/Strep (Thermo Fisher). Cells were grown in a 37 °C incubator with a 5% CO2 environment. Passage number in all experiments ranged from five to ten.
Primary human B165 (MGMT-methylated, male, Generous Gift of Dr. Albert Kim, Washington University in St. Louis) were cultured as spheres in 100 mm uncoated petri dishes (Fisher) using DMEM/F12 Glutamax media (Gibco), supplemented with 1% Pen/Strep (Thermo Fisher), 2% B27 (Miltenyi Biotec), 2.5 µg/mL heparin (Sigma-Aldrich), 20 ng/mL EGF (PeproTech), and 20 ng/mL bFGF (PeproTech). Cells were grown in a 37 °C incubator with a 5% CO2 environment. Passage number in all experiments ranged from six to ten.
BMAL1 knockdown
To knockdown BMAL1 in LN229 cells, a predesigned shRNA plasmid packaged into a lentivirus (vector pLKO.1, viral titer = 106 particles/mL) was obtained from Sigma Aldrich (target sequence: GCAGAATGTCATAGGCAAGTT). Cells were grown in T-25 cm2 flasks for 24 h and incubated for 10 min at 37 °C in 3 mL complete DMEM media with 10% FBS (Thermo Fisher), 5% Pen/Strep (Thermo Fisher), and 15 mg polybrene (Millipore #TR-1003-G). Following incubation, 5–10 mL of virus stock solution was added to each culture. Media was changed after 24 h and cells were kept at 37 °C, 5% CO2. After infection, cells were selected using puromycin (1.5 mg/mL, Thermo Fisher) for ten days. Knockdown efficiency was quantified by qPCR.
Quantitative real-time PCR (qRT-PCR) and quantitative methylation specific PCR (qMSP)
Genomic DNA (gDNA) was extracted and purified from 500,000 cultured LN229, LN229-BMAL1 KD, B165, and Nf1−/−DNp53 GBM cells, synchronized by a media change [11, 17], and collected at CT4 or CT16 using the DNeasy Blood and Tissue Kit (Qiagen), according to the manufacturer’s instructions. The purified gDNA underwent bisulfite conversion using the EZ DNA Methylation-Gold Kit (Zymo Research), following the protocol provided by the manufacturer. The concentration and purity of gDNA was measured at each step using a NanoDrop spectrophotometer (Fisher Scientific). Gene expression changes were further probed using iTaqTM Universal SYBR Green Supermix (Bio-Rad). MGMT-unmethylated and Beta-actin primers, as well as non-converted DNA, were used as internal controls. Universal primers recognizing both methylated and unmethylated Beta-actin sequences were used. The following primer sequences were used: MGMT-methylated forward 5′-TTTCGACGTTCGTAGGTTTTCGC-3′, MGMT-methylated reverse 5′-GCACTCTTCCGAAAACGAAACG-3′, MGMT-unmethylated forward 5′-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3′, MGMT-unmethylated reverse 5′-AACTCCACACTCTTCCAAAAACAAAACA-3′, Beta-actin forward 5′-CTTCGCGGGCGACGAT-3′, and Beta-actin reverse 5′-CCACATAGGAATCCTTCTGACC-3′. qPCR amplification was carried out at 40 cycles with 10 ng of bisulfite treated gDNA in triplicates. Protocol is as follows: Cycle 1: 95 °C for three minutes; Cycle 2: 95 °C for 30 s Cycle 3: 60 °C for 30 s; repeat cycles 1–3 for 39 more times; Cycle 4: 72 °C for 1 min. Negative controls included no template DNA samples. All procedures were done in triplicate in three biological replicates.
Immunostaining and microscopy analysis
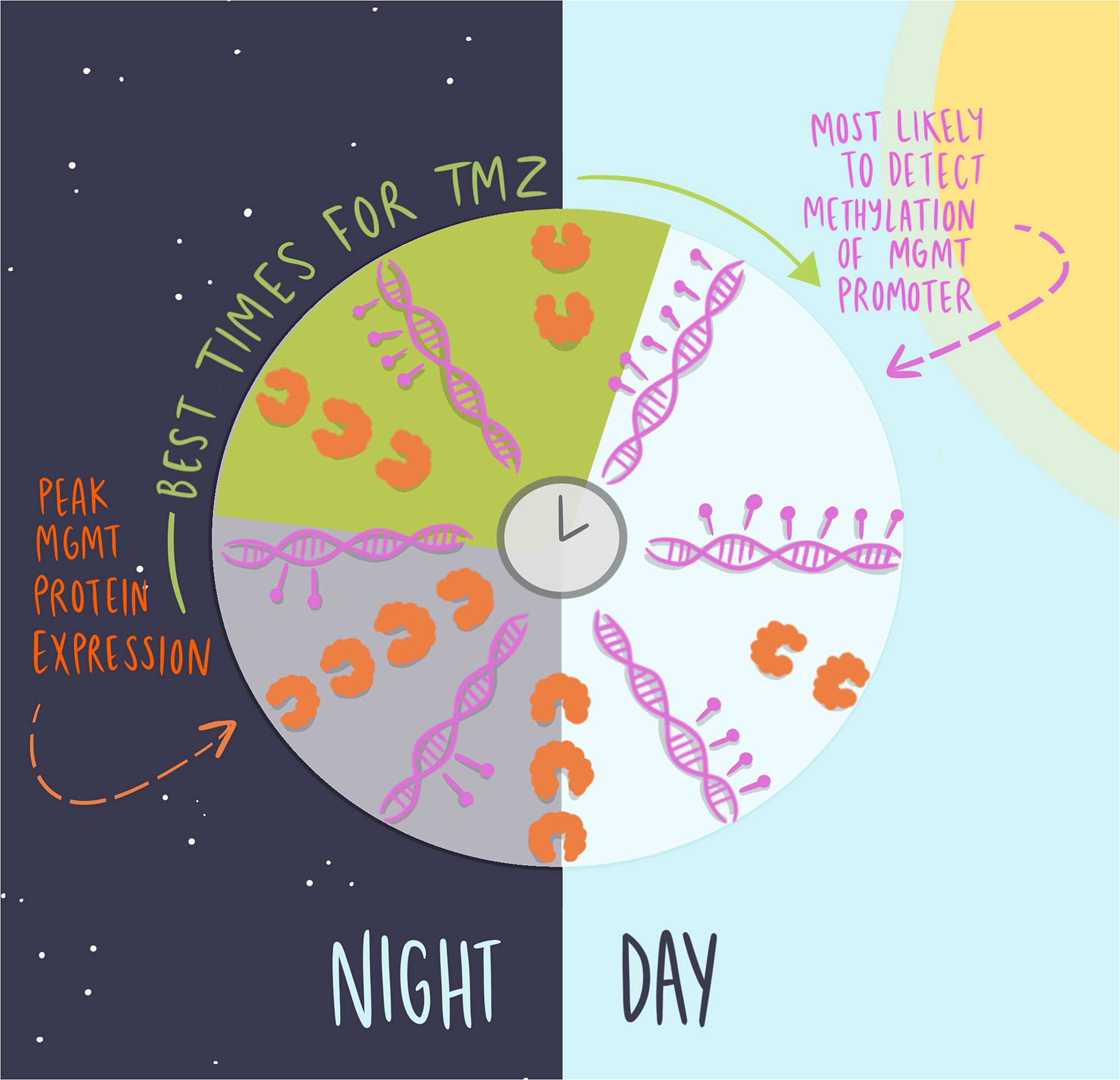
Cells were grown in 35 mm dishes containing a glass coverslip, synchronized by a media change [11, 17], and fixed every 4 h for 24 h, starting 24 h after initial plating, using 4% PFA for 10 min. Circadian time (CT) was defined relative to expression of the clock gene Per2, where CT0 corresponded to peak Per2 expression [11, 17]. Cells were permeabilized for 30 min with 3% Triton-X (Millipore Sigma) in 1x PBS, and blocked for 1 h with solution containing 10% BSA (Sigma) and 0.3% Triton-X. Mouse anti-MGMT (1:500, Invitrogen, Waltham, MA) and rabbit anti-BMAL1 (1:500, Abcam, Cambridge, MA) were diluted in 2% blocking solution and incubated overnight at 4 °C. Samples were then rinsed three times with 1x PBS and incubated in secondary antibody solution (1:500 Alexa 488 goat anti-mouse IgG, and 1:500 Alexa 647 donkey anti-rabbit IgG, Abcam, Cambridge, MA) in 2% blocking solution for 1 h at room temperature. Samples were rinsed three times in PBS, stained with ProLong Gold mounting medium with DAPI (Life Technologies, Carlsbad, CA), and stored in darkness at 4 °C until imaging. To ensure specificity of the primary antibodies binding to the antigen and the secondary antibody binding to the primary, we included control samples with no primary or secondary antibodies, respectively. Each experiment was done in triplicate, in two biological replicates. Six imaging frames were collected and averaged per dish. Microscopy analysis was performed using ImageJ software. Quantification of protein expression was performed as corrected total cell fluorescence (CTCF) = Integrated Density – (Area of selected cell * Mean fluorescence of background readings), as described by McCloy et al., 2014.
In vitro cell growth assays and pharmacology with daily TMZ
GBM cells were plated at the same density (100,000 cells/well of a 6-well plate), synchronized by a media change [11, 17], and grown for 48 h to allow for attachment and growth. Cells were then treated with one of three TMZ concentrations (10, 100, 1000 µM) or vehicle (DMSO, 0.2%), at either Circadian time 4 (CT4) or CT16. Circadian time was defined relative to expression of the clock gene Per2, where CT0 corresponded to peak Per2 expression [11, 17]. Cells were fixed after 72 h with 4% paraformaldehyde (PFA) and stained with 4′,6-diamidino-2-phenylindole (DAPI, 2 mg/mL). We chose to measure survival three days after TMZ administration to allow for approximate two-three cycles of cell division and TMZ-induced DNA lesions, as done previously [11]. DAPI fluorescence was quantified with the Infinite 200 PRO plate reader (V_3.37_07/12_Infinite, Tecan Lifesciences), and fluorescence was converted to cell number using a linear regression equation (Y = 0.009201*X + 2153) obtained from a standard curve where DAPI fluorescence was measured from different number of plated cells. We calculated the percent of cell death as 100 minus percent cell survival (percent cell survival = number of living cells treated with TMZ divided by the number of living cells treated with vehicle). All procedures were done in triplicate in three biological replicates.
Statistical analysis
The Jonckheere-Terpstra-Kendall (JTK) cycle algorithm, a non-parametric test used to distinguish between rhythmic and non-rhythmic data, was used to assess circadian rhythmicity in MGMT protein expression in vitro and the probability of MGMT promoter methylation in patient biopsies. We used p < 0.05 to designate significant circadian rhythmicity [18]. To avoid compromising the assumption of roughly even coverage, we limited our analysis to times when we had 6–15 biopsies. In addition, we applied the Rayleigh statistical test to evaluate differences in the number of patients with GBM scores of methylated or unmethylated as a function of time of day. Statistical significance of mean differences was determined by either Student’s t tests or two-way analysis of variance (two-way ANOVA) with multiple comparisons and Šídák’s post hoc test. All statistical analyses were performed in Prism (version 10.3.0).
Retrospective analysis of MGMT promoter status in GBM biopsies
To determine if MGMT promoter methylation status varied by time of day in human GBM patients, we performed a retrospective chart review on patients diagnosed with GBM at Barnes-Jewish Hospital/Siteman Cancer Center. Inclusion criteria included all patients with newly diagnosed WHO Grade IV GBM (IDH-wildtype), diagnosed from January 2020 to October 2024 to ensure reliable determination of IDH status and MGMT promoter methylation status. A total of 302 patients met these inclusion criteria. Their clinical and demographic variables, such as MGMT promoter methylation status, age, sex, and time of surgery were collected. Time of surgery was determined by the recorded end time of anesthesia to approximate when the tissue samples were embedded for analysis. MGMT methylation status was determined by bisulfite PCR profiling of the MGMT promoter sequence. JTK cycle was used to assess circadian rhythmicity in MGMT promoter methylation status, with a level of p < 0.05 used to designate significant circadian rhythmicity [18].
Modeling methods
To simulate the effects of TMZ on tumor DNA in vitro, the following system of differential equations was used:
$$\frac}} = Z(1 - D) - MD$$
$$\frac}} = - \alpha \sin (t\pi /12)$$
In this cell-based model, \(\:Z\) represented TMZ toxin (MTIC) concentration (with dynamics taken from a previously published model), \(\:M\) represented MGMT protein concentration, and \(\:D\) represented the fraction of double-stranded damaged DNA in tumor cells in vitro [19]. We next chose to compare this model to a simpler model in which TMZ was modeled as an impulse followed by exponential decay (see Supplemental Figures S3-S6). Additionally, exploratory simulations were carried out using a modified equation for double-stranded DNA damage in which a Michaelis-Menten term was added (Supplemental Figures S7-S9), as well as model predictions using a non-sinusoidal daily waveform of MGMT production (Supplemental Figure S10-S12). Because findings did not significantly differ across models, the results from the cell-based model without the Michaelis-Menten term are presented in the main text and figures.
In this system of equations, \(\:Z\:\)created damaged DNA \(\:D\) at rate \(\:_\), in proportion to the amount of undamaged DNA, \(\:1-D\). The parameter \(\:_\:\)removed damaged DNA in proportion to the amount of available MGMT and damaged DNA, \(\:MD\). We adjusted the original model to include circadian variations in \(\:M\) (i.e., MGMT protein) as a sinusoid with a period of 24 h, with initial conditions chosen such that it reached its maximum value 4 h after the start of the simulation, circadian time 4 (CT4), in keeping with previous experimental data. The MGMT derivative scalar \(\:\alpha\:\) and initial condition \(\:_\:\)were chosen to enforce the timing of the maximum MGMT and a 10-fold change in \(\:M\) over the course of the day. Finally, we assumed that the initial conditions for \(\:Z\) and \(\:D\) were \(\:_=0\) and \(\:_=0\), respectively. This reduced our unknown parameters at baseline to \(\:_\:\)and \(\:_\), corresponding to the relative strengths of TMZ at inducing DNA damage and MGMT at repairing it.
Cell death
To model TMZ-induced apoptosis, we assumed that the likelihood of cell death was proportional to the total DNA damage over time; \(\:\int\:D\left(t\right)dt=\varvec\). We also assumed that DNA damage exceeding threshold \(\:_\) killed all cells. This fatal threshold \(\:_\:\)can be solved for uniquely using data that shows that 1000 µM TMZ demonstrates approximately 60% as much variation in dosing efficacy over the course of the 24 h day as 100 µM TMZ [11].
This in vitro model made the following assumptions: (1) The only factor affecting the impact of TMZ is the presence or absence of MGMT, (2) the apoptosis decision is based on cumulative DNA damage (the area under the TMZ-induced damage curve), and (3) the extent of variation of MGMT protein over the course of the day approximately agrees with the variation in the MGMT gene (10-fold difference between maximum and minimum).
To handle the unknowns around the parameters reflecting the relative strengths of TMZ vs. MGMT, we simulated a wide range of potential values. To find optimal dosing times for TMZ, the quality of the model fit was determined using the mean and variation (maximum - minimum) of cell death induced by TMZ across all times of day from published data [11]. By excluding information linking timing to TMZ efficacy in fitting this model, we aimed to predict time-of-day variations in TMZ efficacy based solely on first principles and the MGMT protein data reported here.





Comments (0)